T. KOGA, M. MATSUOKA, K. KITAMURA, N. HIGASHI
Department of Molecular Science and Technology, Faculty of Engineering, Doshisha University, Kyotanabe, Kyoto 610-0321, JAPAN
Tel:+81-774-65-6621, Fax:+81-774-65-6844, E-mail:tkoga/mail.doshisha.ac.jp
The self-assembly of peptides into shape-specific nanostructures has attracted much attention as a powerful approach for the design of novel functional nanomaterials and because of their association with neurodegenerative diseases. We report herein a novel method for the control of peptide self-assembly using synthetic b-sheet peptides composed of l- or d-amino acid as building blocks. Amyloid-like nanofiber formation and its nanostructure could be regulated by the stereospecificity of the constituent peptide species. Triblock-type amphiphilic oligopeptides, 1L and 1D, were designed to have tetra- l-or d-leucine domains, which provide the hydrophobic driving force for self-assembly, flanking pH-responsive l- or d-(lysine)8 segment, respectively. Detailed analyses of the conformation and morphology in aqueous media were performed by using AFM, CD, FTIR measurements. The experimental results revealed that the 1L self-assembled into amyloid-like nanofibers with the conformational transition from a-helix to b-sheet under certain conditions. These nanofibers possessed a diameter of ca. 6.0 nm and a clearly visible left-handed twist with a periodicity of ca. 50 nm. The 1D also showed an a-to-b structural change with similar transition kinetics, but self-assembled into right-handed nanofibers. Thus, the helicity of self-assembled nanofibers could be easily controlled by the chirality of the amino acids in the constituent peptide species. More interestingly, such nanofiber formation was not observed for 1L/1D mixed system, and the racemic mixture formed only globular aggregates. In this case, the peptides 1L and 1D formed co-aggregates rapidly owing to the relatively strong hydrophobic interaction, and the resultant 1L/1D-complex at the initial stage probably prevented the control growth of the peptides required for nanofiber formation.
L. RUBATAT a,b, Z. SHI a, O. DIAT c, S. HOLDCROFT a,d , B.J. FRISKEN a
a Department of Physics, Simon Fraser University, Burnaby, BC, V5A 1S6, Canada (shi/sfu.ca, frisken/sfu.ca, holdcroft/sfu.ca)
b present : Physics Dept. Fribourg University, 1700 Fribourg, Switzerland (laurent.rubatat/unifr.ch)
c UMR SPrAM 5819, CEA-Grenoble, 17 av. des Martyrs, 38054 Grenoble, cedex 9, France (olivier.diat/cea.fr)
d Institute for Fuel Cell Innovation, National Research Council Canada, 3250 East Mall, Vancouver, BC, V6T 1W5, Canada
We use di- and triblock copolymer as a model system to the correlation between morphology and transport properties in ionomer materials. Block copolymers with a fluorinated block and a sulfonated polystyrene blocks allow control of the ionic exchange capacity (IEC) by adjusting either the length of the sulfonated polystyrene chains or their degree of sulfonation.
We have studied the structure of membranes made from various polymer configurations by Small Angle Neutron Scattering (SANS) using contrast variation. Analysis shows that there is phase separation at length scales of the order of a few tens of nanometers due to the non-miscibility of the two polymer blocks; and that there is substructure within the sulfonated polystyrene domains due to separation between the hydrated ionic groups and the hydrophobic polystyrene. We compare these results to TEM images and measurements.
J.-C. PERRIN1, S. LYONNARD1, F. VOLINO2, A. GUILLERMO1
1 UMR 5819 (CEA-CNRS-UJF) DRFMC/SPrAM, CEA Grenoble Cedex 9. Groupe “Polymères Conducteurs Ioniques”
2 UMR 5819 (CEA-CNRS-UJF) DRFMC/SPrAM, CEA Grenoble Cedex 9. Groupe “Théorie”
We have investigated the water dynamics in different ionomer membranes (Nafionâ and sulfonated polyimides) used for fuel cell applications. The membranes exhibit a nanophase separation between the hydrophobic matrice and ionic clusters. The multi-scale hierarchical structure of these porous materials is yet not completely understood. The study of the dynamical behaviour of water molecules from the molecular level to the macroscopic scale as a function of the hydration state of the membrane is of great importance to understand the protonic conductivity mechanism and the relation between macroscopic properties and microstructure.
In order to study the water dynamics in a wide range of
correlation times, we have used complementary techniques
simultaneously: quasi-elastic neutron scattering (QENS), NMR
relaxometry, and pulsed field gradient NMR. The PFG-NMR
experiments allowed to measure the self-diffusion coefficient Ds
of the membranes as a function of the water content. From the
neutron scattering experiments, we extracted the typical
time-scale and length-scale of the molecular motions and showed
that the water mobility increases with water content up to almost
bulk-like behavior. In the hydrated Nafionâ, we have
analysed the QENS data with a Gaussian model for localized
translational diffusion taking into account the existence of
long-range diffusion at the nanometer scale [1]. The typical
confinement size, the local and nanometer diffusion coefficients,
as well as the characteristic times for the elementary jump
process, has been obtained as a function of the water loading and
are discussed with respect to sorption, structural and
self-diffusion properties [2]. At intermediate timescales, the
NMR relaxometry is the most suitable technique to investigate the
proton motion in the range of 20 nanoseconds to 20 microseconds.
The NMR longitudinal relaxation rate R1 measured over
three decades of Larmor angular frequencies w is particularly
sensitive to the host-water interactions and thus well suited to
study fluid dynamics in restricted geometries. In the polyimides
membranes, we have observed a strong dispersion of R1(w)
following closely a ![]() law in a low frequency range (correlation
times from 0.1 to 10 ms). This is indicative of a strong
interaction of water with “interfacial” hydrophilic groups of
the polymeric matrix (wetting situation). Variations of the
relaxation rates with water uptake reveal a two-step hydration
process: solvation and formation of disconnected aqueous clusters
near polar groups, followed by the formation of a continuous
H-bond network. On the contrary, in the Nafionâ we
observed weak variations of R1(w) at low frequency.
This is typical of a non-wetting behavior. At early hydration
stages, R1(w) evolves logarithmically suggesting a
confined bidimensional diffusion of protons in the microsecond
time range. Such an evolution is lost at higher swelling where a
plateau related to 3D diffusion is observed.
law in a low frequency range (correlation
times from 0.1 to 10 ms). This is indicative of a strong
interaction of water with “interfacial” hydrophilic groups of
the polymeric matrix (wetting situation). Variations of the
relaxation rates with water uptake reveal a two-step hydration
process: solvation and formation of disconnected aqueous clusters
near polar groups, followed by the formation of a continuous
H-bond network. On the contrary, in the Nafionâ we
observed weak variations of R1(w) at low frequency.
This is typical of a non-wetting behavior. At early hydration
stages, R1(w) evolves logarithmically suggesting a
confined bidimensional diffusion of protons in the microsecond
time range. Such an evolution is lost at higher swelling where a
plateau related to 3D diffusion is observed.
R. BANSILa, M. LIa, Y. LIUa, H. NIEa, M. STEINHARTc
aPhysics Department, Boston University, Boston, MA 02215, USA (rb/bu.edu)
bAcademy of Sciences of the Czech Republic, Heyrovského nám. 2, CZ-162 06 Praha 6, Czech Republic
Block copolymers and lipids exhibit transitions from
cylindrical micelles to spherical micelles or lamellar (LAM)
phases. The kinetics of the HEX-BCC transition can be described
by coupling anisotropic fluctuations on cylinders, similar to the
pearling instability, according to which the amplitude of a
transverse wave along the length of the cylinder grows causing
the cylinder to break up into spheroidal droplets. We find that
the sphere BCC phase arises with phase shifts of 0, 4π/3
and 8π/3 for the sinusoidal waves on 3 neighboring
cylinders on the HEX lattice, which correspond to the minimum of
overlap volume of rippled cylinders and wavelength λ
related to the nearest neighbor distance of the rippled cylinders
by ![]() . The
azimuthally averaged scattering function from an un-oriented
system of cylinders, as well as the 2-dimensional scattering from
an oriented system was calculated with varying amplitude of the
fluctuation. The results are in excellent agreement with
time-resolved SAXS measurements of the kinetics of this
transition in a Styrene (S)-ethylene-co-butylene (EB)-Styrene
(S)) triblock copolymer in mineral oil, a selective solvent for
the EB block. Similar calculations were also done to describe the
transition from lamellar to HEX phase and compared with SAXS
measurements of kinetics of Lam-HEX transition.
. The
azimuthally averaged scattering function from an un-oriented
system of cylinders, as well as the 2-dimensional scattering from
an oriented system was calculated with varying amplitude of the
fluctuation. The results are in excellent agreement with
time-resolved SAXS measurements of the kinetics of this
transition in a Styrene (S)-ethylene-co-butylene (EB)-Styrene
(S)) triblock copolymer in mineral oil, a selective solvent for
the EB block. Similar calculations were also done to describe the
transition from lamellar to HEX phase and compared with SAXS
measurements of kinetics of Lam-HEX transition.
This research was supported by NSF-DMR.
D. BENDEJACQa, V. PONSINETb
a Centre de Recherche d’Aubervilliers, 52 Rue de la Haie Coq 93308 Aubervilliers Cedex, France
b Centre de Recherche Paul Pascal, CNRS UPR8641, Ave. Schweitzer, 33600 Pessac, France (ponsinet/crpp-bordeaux.cnrs.fr)
We investigate the self-assembly of poly(styrene-stat-(acrylic acid))-block-poly(acrylic acid), i.e., P(S-stat-AA)-b-PAA, diblock copolymers. We observe that as the composition of the statistical first block is varied, these diblocks form, in the melt, strongly to weakly microphase-separated structures, or homogeneous (disordered) phases. We thus demonstrate that the segregation strength can be quantitatively controlled by the composition of the statistical first block, regardless of the diblock symmetry. We show by small-angle X-ray and neutron scattering that the structural behaviour in water of these diblocks is controlled by the amount of acid segments in the first block. For low amounts, the structures found in water are frozen micelle-like objects, reminiscent of the macro-phase separation in the solid state. For high amounts, the diblocks are soluble in water. In the intermediate regime, an equilibrium self-association occurs, which present variable morphologies and is controlled by the pH and the ionic strength of the dispersion. We discuss the partition of the charges between the two polyelectrolyte blocks to account for this behaviour.
O.V. BORISOV
University of Pau, 2, Av. President Angot, 64053 Pau, France, borisov/univ-pau.fr
Theory of self-assembly in water-based copolymer systems controlled by balance of ionic, steric and hydrophobic interactions is presented.
We discuss first the effect of intra-molecular hydrophilic-hydrophobic balance on structure and morphology of micelles of diblock copolymers with ionic and hydrophobic blocks. Phase diagrams of the system including regions of stability of generic morphologies and co-existence lines are constructed. In particular, we focus on copolymers comprising, e.g., pH and thermo-sensitive building blocks capable to form aggregates with stimuli-responsive properties. We demonstrate, that variation in pH or salinity of the solution can trigger abrupt structural re-arrangements of the copolymer aggregates, that is a manifestation of coupling between ionisation and aggregation of the macromolecules. Furthermore, the effects of progressively increasing topological complexity of amphiphilic macromolecules (graft-copolymers, core-shell architectures) on the routes of self-organization, polymorphism of aggregates, balance of intra- and inter-molecular association are considered. Nature of stimuli-induced abrupt morphological and conformational transitions versus continuous transformations is discussed in the context of design of intelligent nano-structures tailored for specific response to controlled variation of the environmental conditions. Predictions of analytical theory based on statistico-thermodynamic approach are systematically compared to the numerical modelling and experimental results.
S. HVIDT
Department of Chemistry, Roskilde University, Universitetsvej 1, DK-4000 Roskilde, Denmark (hvidt/ruc.dk)
Triblock copolymers of the type EPE, where E and P denote ethylene oxide and propylene oxide blocks, respectively, are industrially important copolymers called Pluronics or Poloxamers. EPE copolymers form micelles with a core of P blocks and different micellar shapes depending on block length ratios and temperature. The micellization process with increasing temperature has been followed by a number of techniques including differential scanning calorimetry, liquid chromatography, and surface tension measurements. These results are not in agreement with the simple classical model for micellization but indicate a more gradual formation of micelles, which have often been interpreted as due to formation of small copolymer aggregates prior to micellization. Recent studies [1,2] have shown, however, that chemical heterogeniety of the copolymers also has an effect on micellization. Different micellization models have been tested on purified copolymers and copolymer mixtures, and evidence in favor of a multi-equilibria unimer-micelle model will be presented.
EGPC is a liquid chromatographic method, which can be used to investigate dynamics of unimer-micelle systems [3] and also the formation of mixed micelles of two copolymers. It will be shown that Poloxamers contain from 10-24% impurities by mass, which are not incorporated into micelles. The presence of these impurities is due to a chain-transfer reaction during synthesis. The micellization of intact tri-block copolymers occurs over a wide temperature interval, as seen in both DSC and EGPC. The width of the transition is primarily due to a distribution of PPO blocks, and long blocks result in micellization at low temperatures. The use of the EGPC technique for studies of mixed micelle formation will finally be demonstrated.
[1] Hvidt, S., Trandum, C., Batsberg, W. (2002) J. Colloid Interface Sci. 250, 243.
[2] Batsberg, W., Ndoni, S., Trandum, C., Hvidt, S. (2004) Macromolecules 37, 2965.
[3] Nixon, S.K., Hvidt, S., Booth, C. (2004) J. Colloid Interface Sci. 280, 219.
M. MÜLLERa, W. OUYANGa, B. KEßLERa
aLeibniz-Institute of Polymer Research Dresden e. V. (IPF), Hohe Str. 6, 01069 Dresden
Polyelectrolyte (PEL) complexes (PEC) are self-assembled systems and can be prepared by two concepts (A, B): mixing oppositely charged PEL in solution leads to dispersed nanoparticles (Bungenberg de Jong, Kabanov and Dautzenberg) (A), consecutively adsorbing those PELs at solid interfaces results in PEL multilayer (PEM) films (Decher, Möhwald) (B). Varying the PEL stiffness it will be pointed out that PEC and PEM show significant correlations in the adopted nanostructure, although the preparation is apparently different. Charged homopolypeptides like cationic poly(L-lysine) (PLL) and anionic poly(L-glutamic acid) (PLG) were used, whose conformations can be changed from random coil to a-helix by salt type or pH value [1-3]. PLL and PLG were combined with certain polyanions (poly(vinylsulfate) (PVS), poly(maleic acid-co-alkenes) (PMA-X)) and polycations (PDADMAC, PEI), respectively. in-situ ATR-FTIR, AFM, circular dichroism (CD) and dynamic light scattering (DLS) were used for characterization.
 a) a) |
 b) b) |
 c) c) |
 d) d) |
| Fig. 1. [2-3] AFM images of PEC of PLL/PMA-MS (pH=6) (a), PEM of PLL/PVS (pH=6) (b) containing randomly coiled PLL and PEC PLL/PMA-P (pH=6) (c), PEM of PLL/PVS (NaClO4) containing a-helical PLL (d). | |||
Fig. 1 shows AFM images of casted PEC particle dispersions and consecutively adsorbed PEM films containing PLL either in the random coil (a, b) or a-helical conformation (c, d) leading to isotropic or anisotropic nanostructures, respecticvely [2-3].
[1] M. Müller, B. Keßler, K. Lunkwitz, J. Phys. Chem., 107 (32), 8189-8197 (2003)
[2] M. Müller, T. Reihs, B. Keßler, H.J. Adler, K. Lunkwitz, Macromol.Rapid. Commun., 25(3), F56-F57 (2004)
[3] M. Müller, T. Reihs, W. Ouyang, Langmuir, 21(1), 465 (2005)
M. BURKHARDT1, M. RUPPEL1, N. MARTINEZ-CASTRO1, S. TEA1, D. PERGUSHOV2, M. GRADZIELSKI3, A.H.E. MÜLLER1;
1Makromolekulare Chemie II, Universität Bayreuth, Germany,
2Moscow State University, Russia, 3Stranski-Laboratorium für Physikalische und Theoretische Chemie, TU Berlin
Up to now only few investigations of dynamics of block copolymer systems have been reported. Mostly copolymers with a hydrophobic block with Tg above room temperature have been investigated (PS-PAA, PS-PMAA,…). Here the dynamic processes of the aqueous solutions are hindered due to the glassy state of the PS core of the micelle. In our case we employ polyisobutylene (PIB) as the core-forming block, that should exist in a liquid-like state (Tg ≈ -55°C). Therefore changes in the solution parameters (pH, salt concentration) could have an effect not only on the corona, but also on the core of the micelle, i.e. one expects a dynamic behaviour of such aggregates. This should also have an effect on the aggregation number.
Also Interpolyelectrolyte Complexes (IPECs), formed by the polyelectrolyte in the corona of the micelle - poly(methacrylic acid) (PMAA) - with quaternized poly(4-vinylpyridine) (P4VPQ) have been investigated with respect to their kinetics. At pH = 7, water-soluble complexes are formed up to a Z -value (Z = [P4VPQ]/[PMAA], the basemolar concentrations of the cationic polyelectrolyte and ionic block of the copolymer are given in brackets) of 0.4 - 0.5; beyond this value precipitation occurs. Their complexes are stable at low salt concentrations. However, beyond a certain NaCl concentration, the complexes dissolve, thereby reforming the originally present copolymer micelles and molecularly dissolved polyelectrolyte. This can be proven by different types of measurements, including SANS, turbidimetric titration, titration with a sodium-selective electrode and stopped-flow in combination with SAXS. Kinetic investigations of the formation and the dissociation of the complex show an overlay of a fast mixing process, followed by a slower process. The faster one is believed to be the diffusion of individual copolymer molecules coupled to the diffusion of the polycation and the salt into or out of the micelle. The slower one could be a kind of dynamic rearrangement of the block copolymers to form the finally stable aggregates.
Thermodynamic investigations on the enthalpy of formation of the IPEC as well as thermodynamic data from titrations are planned to give a deeper insight into the processes occurring during formation and salt-induced dissociation of such complex macromolecular assemblies.
Literature:
1.) D. Pergushov, E. Remizova, J.Feldthusen, A. Zezin, A.H.E. Müller, V. Kabanov; Novel Water-Soluble Micellar Interpolyelectrolyte Complexes, J. Phys. Chem. B, 2003, 107, 8093-8096
2.) D. Pergushov, E. Remizova, M. Gradzielski, P. Lindner, J. Feldthusen, A. Zezina, A.H.E. Müllere, V. Kabanov; Micelles of polyisobutylene-block-poly(methacrylic acid) diblock copolymers and their water-soluble interpolyelectrolyte complexes formed with quaternized poly(4-vinylpyridine); Polymer, 2004, 45, 367-378
N. KARANIKOLOPOULOS, M. PITSIKALIS, N. HADJICHRISTIDIS
Department of Chemistry, University of Athens, Panepistimiopolis Zografou, 15771 Athens, Greece, (pitsikalis/chem.uoa.gr, http://www.chem.uoa.gr/courses/polymers/PG_Polym_Index.htm )
Drug delivery systems have attracted increasing attention the last two decades. Among drug carriers micelles provide a set of unbeatable advantages. In this study PEO-b-PDMAEMA block copolymers, where PEO is poly(ethylene oxide) and PDMAEMA poly(dimethylaminoethyl methacrylate) were prepared by atom transfer radical polymerization. Part of the dimethylamine groups was transformed to zwitterions by reaction with 2-chloro-2-oxo-1,3,2-dioxophospholane. The micellization behavior was studied in water by light scattering techniques. Ether phospholipids, which have been shown to be very active for the treatment of Leishmania, were incorporated in the micellar structures leading to the establishment of equilibrium between micelles and vesicles. The effect of the copolymer molecular weight and composition, the percentage of the zwitterionic groups, the solution pH and temperature on the micellization behavior was examined.
K.I. WINEY
University of Pennsylvania, Material Science and Engineering, 3231 Walnut St., Philadelphia, PA 19104-6272, USA, winey/seas.upenn.edu
We have demonstrated the ability to detect ionic aggregates in ionomers using high angle annular dark field scanning transmission electron microscopy (HAADF STEM). This microscopy method minimizes phase contrast, which dominates TEM images at high magnification (typically 1,000,000X), and accentuates the atomic number contrast between the hydrocarbon polymer and the metal cations. We have applied this tool to a variety of ionomer systems and along with the anticipated spherical aggregates we have also found vesicular and rod-like aggregates and macrophase separation. We have also identified processing methods that alter the state of ionic aggregation and aggregate shape in PDMS-based and PS-based ionomers.
Our current focus is on rigorously testing the small angle x-ray scattering (SAXS) model proposed by Yarusso and Cooper for the interaggregate scattering in ionomers. Preliminary studies have been completed using model nanoparticles (11 and 55 atom Au clusters) in which the sizes determined by STEM and SAXS are in agreement. Efforts are currently underway to quantitatively compare results from STEM image for the size and number density of ionic aggregates with SAXS interpreted by the Yarusso-Cooper model. The model materials for this study are poly(styrene-ran-methacrylic acid) copolymers fully neutralized with copper.
V.V. Khutoryanskiy
School of Pharmacy, University of Reading, Whiteknights, PO Box 224, Reading RG6 6AD Berkshire, United Kingdom. E-mail: v.khutoryanskiy/reading.ac.uk
Water-soluble non-ionic polymers are able to form hydrogen-bonded interpolymer complexes with poly(carboxylic acids) in aqueous solutions. This topic in polymer research has received much attention since sixties. In the present communication the recent progress in the field of complexation and blending of polycarboxylic acids with various water-soluble non-ionic polymers via hydrogen bonding will be considered. The following topics will be highlighted:
References
H. MENDILa, P. BARONIa, L. NOIREZa
Laboratoire Léon Brillouin (CEA-CNRS), Ce-Saclay, 91191 Gif-sur-Yvette Cédex, France (noirez/llb.saclay.cea.fr)
Flow instabilities as the shear induced phase transitions (fig.1) are some of the most spectacular but also less explained phenomena[1]. The origin of these very spectacular non-linear behaviours is unknown. We claim that the key parameter is this debate is the existence of a primary low frequency elastic mode which has been first identified in side-chain liquid crystal polymers and later in ordinary polymer melts[2,3]. These novel experimental observations imply in particular that the flow regime is not the terminal behaviour of molten polymers, even at temperatures far away from the glass transition. The existence of this additional low frequency mode shows that a so far unknown intrinsic polymer blend property has been neglected and should be taken into account to describe adequately various so far unexplained shear induced phenomena. We illustrate our purpose by determining and comparing the characteristic times of an ordinary polymer and a LC-polymer system to the critical times associated with the shear induced instabilities.

1.C. Pujolle-Robic, L. Noirez, Nature 409 (2001) 167.
2. D. Collin, P. Martinoty, Physica A 320 (2002) 235.
3. H. Mendil, P. Baroni, L. Noirez, Eur. Phys. J. E 19 (2006) 77
P. PANINEa, N. DIDIERb
aEuropean Synchrotron Radiation Facility, B.P. 220, 38043 Grenoble, France (panine/esrf.fr)
bEcole Nationale Supérieure de Physique de Grenoble, 38402 St-Martin d'Hères Cedex, France (nicolas.didier/grenoble.cnrs.fr)
One of the most important evolution in the development of new class of materials is the way process engineering takes advantage not only from the molecular structure but also from its supramolecular organization. For instance, the key advantage of block copolymers over statistic copolymers resides in the phase separation phenomenon and related order-disorder transition. The large domains appearing during this phase separation provide interesting and often contradictory properties to the final system e.g. soft touch but with high stiffness, separated glass transitions, etc. We are presenting here the shear induced phase transition on a microphase separated block copolymer using a rheometer, coupled on-line to structural investigation by small angle x-ray scattering (SAXS). The development of a new lamellar phase appearing under shear as well as its orientation will be demonstrated to depend on the shear rate. Providing a strict control on the thermodynamical conditions and of the hydrodynamical field during deposition, large mono-oriented domains in polymeric films can be tailored. Quantitative analysis of the distribution of orientation is described.
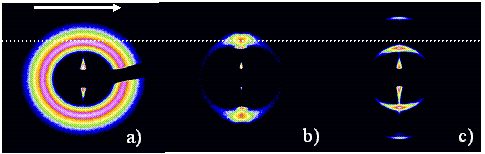 |
Fig. 1
Figure 1: Typical 2D X-Ray SAXS pattern of a shear induced phase transition from a) isotropic short lamellar spacing to c) oriented larger lamellar spacing. b) shows the coexistence. The shear geometry is a cone/plate. Arrow indicates the shear direction. The dotted line is a guide for the eye.
M. GEOGHEGAN,*a L. RUIZ-PÉREZ,a R. LA SPINA,a M.R. TOMLINSON,a C. C. DANG,a A.J. PARNELL,a S.J. MARTIN,a J.R. HOWSE,a R.A.L. JONES,a P. D. TOPHAM,b C.J. CROOK,b A.J. RYAN,b D.S. SIVIA,c J.R.P. WEBSTER,c A. MENELLEd
aDepartment of Physics and Astronomy, Hicks Building, University of Sheffield, Sheffield S3 7RH, United Kingdom
bDepartment of Chemistry, Dainton Building, Univcrsity of Sheffield, Sheffield S3 7HF, United Kingdom
cISIS Pulsed Neutron and Muon Source, Rutherford Appleton Laboratory, Chilton, Didcot, Oxfordshire OX11 0QX, United Kingdom
dLaboratoire Léon Brillouin, CEA-Saclay (CEA-CNRS), F-91191 Gif-sur-Yvette Cédex, France
Polymers attached to surfaces (brushes) present a useful means of compatibilisation between organic and inorganic surfaces. However the conformation of polyelectrolyte brushes represents an interesting problem in polymer physics and is not wholly understood. We present neutron reflectometry data concerning the conformation of polyelectrolyte brushes in an aqueous environment, which suggests that the behaviour of even model polyelectrolytes depends on parameters such as synthetic route. These polyelectrolyte brushes are synthesised by atom transfer radical polymerization, which enables the creation of rather dense brush layers. Despite being dense, it is observed that polybase brushes of poly[(diethyl amino)ethyl methacrylate] (PDEAEMA) form an unexpected conformation in acid solution whereby the volume fraction (concentration) of the brush does not decrease monotonically with depth from the substrate, but rather has a depletion zone. The interaction of polyelectrolyte brushes with polyelectrolyte networks is an interesting combination as it creates the possibility of reversible adhesion, and some results will be presented including the interaction between polymethacrylic acid gels and brushes and PDEAEMA gels with polymethacrylic acid brushes. The polyacid-polybase system appears to be more promising for switchable adhesion than the polyacid-polyacid system in this respect, and these conclusions will be discussed.
N. CLARKE, L.R. HUTCHINGS, A. PILLAY-NARRAINEN, I.A. ANSARI, R.L. THOMPSON
Department of Chemistry, University of Durham, Science Site, South Road, Durham, DH1 3LE, U.K. (r.l.thompson/dur.ac.uk)
We have synthesized a series of linear deuterio-polystyrene (dPS) chains from fluorocarbon functionalised dendritic initiators1 (see figure 1). These polymers, denoted FxdPSy, where x is the number of C8F17 groups and y is the Mn of the dPS (kg/mol) are highly surface-active in blends with unfunctionalised polystyrene. Nuclear reaction analysis (see figure 2) shows spontaneous surface excess formation within the few seconds of the spin-coating process. This indicates that these materials have an appreciable surface-activity, even in solutions. (This is not normally observed for polymers bearing a single fluorocarbon unit2). Increasing the size or the number of the fluorocarbon groups per polymer chain shifts the equilibrium in favour of adsorption such that saturation of the surface layer can be obtained at reduced bulk concentration. Contact angle analysis with both dodecane and water indicates a substantial (~50%) reduction in surface energy as saturation of the surface is approached. The relationship between contact angle and surface excess data will be discussed in the paper, along with preliminary results from neutron reflectometry and small-angle neutron scattering.
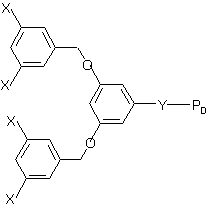
FIG 1. Sketch of F4dPSy dendron functionalised polymer chain.
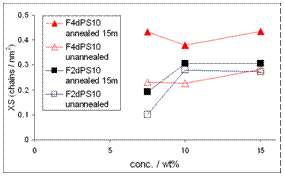

FIG 2. Surface excesses of FxdPS10 blended films. The lack of concentration dependence in the more F4dPS10 indicates surface saturation.
(1) Narrainen, A. P.; Hutchings, L. R.; Ansari, I. A.; Clarke, N.; Thompson, R. L. Soft Matter 2006, 2, 126-128.
(2) Kiff, F. T.; Richards, R. W.; Thompson, R. L. Langmuir 2004, 20, 4465-4470.
J. LAL
Intense Pulsed Neutron Source, Argonne National Laboratory, Argonne, IL, 60439, USA
The interaction of polymers with flexible surfactant layers may modify strongly the elasticity and fluctuations of the layers. This effect has been studied in two surfactant mesophases. Neutron Spin Echo (NSE) spectroscopy was used to probe the modification of membrane fluctuations first in the case of dilute microemulsion droplets both in the presence and absence of polymer. A second example studied more recently, was hydrophobically modified polymers (polysoaps) interacting with fluctuating lamellar bilayers of a nonionic surfactant.
G. LARUELLE a, O.V. BORISOV a, A. LAPP b, J. FRANCOIS a, L. BILLON b
a Laboratoire de Physico-Chimie des Polymères UMR 5067, Université de Pau et Pays de l'Adour, Hélioparc, 2 Avenue Angot, 64053 Pau, France (laurent.billon/univ-pau.fr)
bLaboratoire Léon Brillouin, CEA/CNRS, 91191 Gif-sur-Yvette, France
Two different types of amphiphilic di-block copolymers have been synthesized by direct Nitroxide-Mediated Polymerization NMP. The first one is constituted by a hydrophilic block (polyacrylic acid PAA) and a hydrophobic block (polystyrene PS), using the PS block as a macro-initiator. The proposed method can lead to samples in broad range of composition and molar mass. The second type of copolymer was presented an original macromolecular architecture. Indeed in this case, a block copolymer, where the first block is a pure PAA whereas the second block is PAA-grad-PS was elaborated. The gradient composition of these copolymers have been estimated by 1H and 13C NMR.
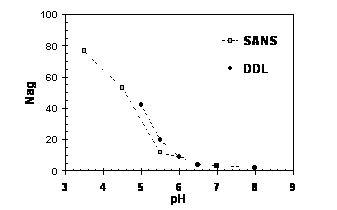 |
These copolymers are directly soluble in pure water at room temperature without special experimental procedure (addition of electrolytes or effect of temperature) and studies of their self-assembly in aqueous medium have been performed. The self-assembly of these amphiphilic block copolymers have been studied by fluorescence and Small Angle Neutron Scattering in order to estimate the Critical Aggregation Concentration CAC and the aggregation number Nagg. Theses copolymers exhibit a correlation peak characteristic of a liquid order organization confirming the formation of micelles with a repulsive corona. Moreover PAA-b-(PAA-grad-PS) amphiphilic block copolymer able to self-assemble in aqueous solution and the strong pH sensitive behaviour of this new type of architecture will be presented.
Variation of the Nagg values as function of pH.
I.I. POTEMKIN, N. OSKOLKOV
Physics Department, Moscow State University, Moscow, Leninskie Gory, 119992 Russia, igor/polly.phys.msu.ru
The effect of spontaneous overcharging of macroions by oppositely charged polyelectrolytes has attracted considerable attention of many theoreticians during the last few years [1-4]. The practical importance of these studies is connected primarily with the understanding of the physical reasons for the DNA-histone complexation [5]. That is why most of the models developed deal with the highly charged polyelectrolytes interacting with oppositely charged hard, impenetrable spheres or cylinders. On the other hand, many macroions, which are "penetrable" for oppositely charged polyelectrolytes, also exhibit overcharging. The examples are the complexes of micelles having polyelectrolyte shell with oppositely charged linear polyelectrolytes [6, 7]. Also microgel particles, star-, comb-like macromolecules, etc., can be classified as penetrable macroions.
The objective of the present study is to demonstrate the mechanism responsible for the overcharging of penetrable macroions in dilute solution [8]. The system of weakly charged polyanion (microgel) and polycations (multi-arm stars) is considered. The effect of positively and negatively charged monovalent counterions is explicitly taken into account. It is shown that the inversion of the charge of the large polyanion (PA) by smaller polycations (PCs), which are able to enter into the PA, is always driven by Coulomb interactions. Counterions can play a dual role: they promote overcharging, if the number of excess PCs is small enough, and prevent overcharging otherwise.
REFERENCES
[1] Gurovitch E. and Sens P., Phys. Rev. Lett., 82 (1999) 339.
[2] Mateescu E. M., Jeppesen C. and Pincus P., Europhys. Lett., 46 (1999) 493.
[3] Park S. Y., Bruinsma R. F. and Gelbart W. M., Europhys. Lett., 46 (1999) 454.
[4] Nguyen T. T., Grosberg A. Yu. and Shklovskii B. I., Phys. Rev. Lett., 85 (2000) 1568.
[5] Rädler J. O., Koltover I., Salditt T. and Safinya C. R., Science, 275 (1997) 810.
[6] Talingting M. R., Voigt U., Munk P. and Webber S. E., Macromolecules, 33 (2000) 9612.
[7] Laguecir A., Stoll S., Kirton G. and Dubin P. L., J. Phys. Chem. B, 107 (2003) 8056.
[8] Potemkin I. I., Europhys. Lett., 68 (2004) 487.
G. TRIMMELa, K. STUBENRAUCHa, A. LEXa, C. MOITZIb, G. FRITZb
aInstitute for Chemistry and Technology of Organic Materials, Graz University of Technology, Stremayrgasse 16, 8010 Graz, Austria
bPhysical Chemistry, Institute of Chemistry, Karl Franzens University, Heinrichstrasse 28, 8010 Graz, Austria
Design of highly ordered and nanostructured materials is one of the most challenging tasks in material chemistry. In this context self-organization behavior of well defined amphiphilic block-copolymers (BCPs) has gained attraction for synthesis of highly functional and hierarchical polymeric materials. Combining different polarities and functionalities into BCPs requires versatile polymerization methods with a high group tolerance. This can be realized by ring opening metathesis polymerization (ROMP) with Grubbs type Ru initiators RuCl2(PCy3)2(CHPh) (1) and. RuCl2(H2IMes)(3-bromo-pyridine)2(CHPh) (2). Especially the latter one has an enhanced reactivity and a higher group tolerance than most other initiators. We exploited the possibilities of 1 and 2 towards the synthesis of AB-BCPs coupling hydrophobic and hydrophilic blocks, side chain liquid crystalline and photo-switchable blocks, semi-perfluorinated and hydrocarbon blocks.
Each combination needs proper choice of solvents and reaction conditions. For example, we prepared amphiphilic block copolymers using as hydrophobic blocks norbornene-dicarboxyclic-acid-dimetylester; and as the hydrophilic moiety norbornene dicarboxylic acid. To guarantee a reproducible synthesis and characterization the acid functionalities were protected as tert-butylesters.
Their abilities of forming micelles in solution as well as their self assembling behaviour in bulk were investigated by scattering techniques (Small Angle X-ray Scattering, Dynamic light scattering) and by electron microscopy. Self assembly of such polymers were investigated for the preparation of highly ordered inorganic-organic hybrid materials.
G. MOUNTRICHAS, S. PISPAS*
Theoretical and Physical Chemistry Institute, National Hellenic Research Foundation, 48 Vass. Constantinou Ave., Athens 11635, Greece (pispas/eie.gr, http://www.eie.gr/nhrf/institutes/tpci/index-en.html )
The synthesis of well-defined double hydrophilic block copolymers via anionic polymerization techniques and post-polymerization functionalization routes is presented. Newly developed model copolymers include sodium poly[(sulfamate-carboxylate)isoprene]-block-poly(ethylene oxide), poly(p-hydroxystyrene)-block-poly(methacrylic acid) and poly[3,5-bis(dimethylaminomethyl)-p-hydroxystyrene]-block-poly(ethylene oxide).
The aqueous solution behavior of the novel block copolymers is found to depend on solution pH and ionic strength, giving rise to the formation of responsive micelles and aggregates. The presence of designed polyelectrolyte blocks makes possible the complexation of the block copolymers with proteins and DNA, allowing for the preparation of functional nanostructures with biotechnological applications potential. The self-assembled systems are studied by different physicochemical techniques, giving information on their structure and self-organization properties, as a function of several parameters of the mixed systems.
P. ŠTĚPÁNEK1, Z. TUZAR1, P. ČERNOCH1, F. NALLET2, L. NOIREZ3
1Institute of Macromolecular Chemistry, Heyrovsky Sq. 2, 162 06 Prague 6, Czech Republic 2Centre de Recherches Paul Pascal, 33 600 Pessac, France, 3Laboratoire Leo Brillouin, CEA Saclay, 91191 Gif-sur-Yvette, France
Mixed diblock copolymer systems will be discussed that contain, besides the diblock copolymer also two immiscible homopolymers or two solvents or their combination. Experimental information is obtained mostly by scattering experiments (light, neutron, X-rays) and atomic force microscopy.
When a diblock copolymer is added to a mixture of two partially miscible solvents (e.g., cyclohexane and dimethylformamide), a nanostructured ordered system is created even for extremely low polymer volume fractions, of the order of a few per cent. The diblock copolymer then covers the interface between the regions of microphase separated solvents. The morphology of the ordered structure observed is generally hexagonal or cubic; the transition from the disordered to periodically ordered state is found to cover an unusually large temperature interval of about 15 degrees above Tc. Macroscopically oriented samples can be created under by application of a shear field.
Dynamic properties of these multicomponent polymer systems have been studied by viscometry, dynamic light scattering and pulsed-field gradient NMR. Various relaxation processes have been described, in particular fluctuations in composition, self-diffusion of the individual components of the system and thermal diffusion. The viscosity of the polymer/immiscible solvents mixture was found to increase by several orders of magnitude during the transition from the disordered phase above Tc into the ordered phase.
When two homopolymers A, B are added to a diblock copolymer A-B either a swollen periodic structure is created or a bicontinuous microemulsion, depending on the relative proportion of the copolymer and homopolymers. For polymers of molar mass larger than a few thousand such structure can be created only in the presence of a solvent that is thermodynamically good for all polymer components of the system. The surface structure of microemulsions studied by atomic force microscopy shows a regular pattern on the scale of a few hundreds nanometers.
We acknowledge support by the Grant Agency of the Czech Republic (SON/03/E001) within the EUROCORES Programme SONS of the European Science Foundation, which is also supported by the European Commission, Sixth Framework Programme.
J. DE JONG, L. LUCAS, R. HANIA, A. PUGLYS, R. KELLOGG, B. FERINGA, J. VAN ESCH*
Stratingh Institute, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands; J.H.van.Esch/rug.nl
The controlled self-assembly of small functional molecules into supramolecular structures is a powerful approach towards the construction of novel devices of nanoscale dimensions. In principle, the structure and properties of supramolecular assemblies depend on the properties of the individual molecules as well as the strength and spatial arrangement of the intermolecular interactions. If the individual molecules contain an addressable function, e.g. are redox, photochromic active or chemoselective, this offers the possibility to change the properties of the supramolecular arrays or the extend of self-assembly by means of an external stimuli.
Hydrogen-bonding motives are because of their high selectivity and directionality especially suited for the design novel molecular building blocks. Here we report on the design and function of new responsive molecular gelators, which are obtained by extension of amide-based gelator scaffolds with addressable groups appended with amides. Self-assembly of photo-responsive gelators based on bis-thienyl photochromic switches has a dramatic influence on the diastereoselectivity of the photochemical ring-closure reaction, and the gelation ability can be turned on and off by photochemical switching between the open and closed form. It was found that these gels can exist in 4 different states pointing to a supramolecular memory effect. Irradiation through a mask or with an interference pattern leads to the reversible formation of spatially confined gel objects. Most remarkably, dynamic pattern formation involving light-induced mass-transfer has been achieved by 2-wavelength irradiation!
1. J. van Esch, B.L. Feringa, Angew. Chem, 2000, 39, 2263-2266.
2. J.J.D. de Jong, L.N. Lucas, R.M. Kellogg, J. van Esch, B.L. Feringa, Science 2004, 304, 278.
3. A. Heeres, C. van der Pol, M. Stuart, A. Friggeri, B.L. Feringa, J. van Esch, J. Am. Chem. Soc. 2003, 125, 14252-14253.
4. K.J.C. van Bommel, C. van der Pol, I. Muizebelt, A. Friggeri, A. Heeres, A. Meetsma, B L. Feringa, J. van Esch, Angew. Chem. 2004, 116, 1695 -1699.
5. J.J.D. de Jong, P.R. Hania, A. Pugzlys, L.N. Lucas, M. de Loos, R.M. Kellogg, B.L. Feringa, K. Duppen,. J.H. van Esch, Angew. Chem. Int. Ed. 2005, 44, 2373 -2376
R.J. Composto
Materials Science and Engineering, University of Pennsylvania, 3231 Walnut St. Philadelphia, PA, 19104-6272, USA, composto/seas.upenn.edu
Self-assembly in polymer films can produce nano to micro-scale structures that have potential use as optoelectronic devices, sensors, microactuators and nanoreactors. In this talk, novel structures are obtained by controlling the thermodynamics and phase evolution pathway of (1) polymer blends containing nanoparticles (NP), (2) in situ NP formation in polymers and (3) amphiphilic block copolymers. The surface composition of NP is found to control NP location in a phase separated blend.1 Interface active NP are found to stabilize 3D interpenetrating or 2D discrete structures depending on the NP volume fraction. A simple interfacial energy argument predicts the conditions for NP jamming at the interface. Interfacially active NPs are also shown to stabilize films. Silver NPs are synthesized in-situ by thermal decomposition in homopolymer and block copolymer films. Surface segregation is observed as well as partitioning of the NPs according to size. Finally, we present a novel route to assemble perpendicular cylinders in block copolymer films by converting poly(styrene-b-tert butyl acrylate) (PS-b-PtBA) to poly(S-b-acrylic acid) (PS-b-PAA) using an auto-catalytic reaction.2 Upon exposure to water, PAA cylinders, constrained by the PS phase, protrude above the surface and swell laterally to form mushroom caps, rendering the entire surface hydrophilic. The swelling-shrinking process is reversible. The nanostructure of PS-b-PAA films also vary with pH. At low pH, PAA chains collapse within the cylinders, resulting in a hexagonal packed array of holes. At intermediate pH, the cylinders swell and transform into mushrooms with a swollen cap. At high pH, PAA chains stretch strongly to cover the entire surface, leading to a continuous PAA wetting layer decorated by hexagonally packed depressions.
Acknowledgements: To the National Science Foundation for funding and H-J Chung, R. Deshmukh and C. Xu for their skillful research.
1 H-J. Chung et al., "Self Regulated Structures in Nanocomposites by Directed Nanoparticle Assembly," Nano Letters, 5, 1878-1882 (2005).
2 C. Xu et al. "Reversible Stimuli-Responsive Nanostructures Assembled from Amphiphilic Block Copolymers," Nano Letters, 6, 282-287, 2006.
M. STEINHART a, P. GÖRINGa, H. DERNAIKAa,b, M. PRABHAKARANa,b, U. GÖSELEa, E. HEMPELb, T. THURN-ALBRECHTb
aMax Planck Institute of Microstructure Physics, Weinberg 2, D-06120 Halle, Germany
bDepartment of Physics, Martin Luther University, Hoher Weg 8, D-06120 Halle, Germany
Polymeric nanorods and nanotubes with a typical diameter of several tens to several hundreds of nanometers can be prepared by wetting of templates with oriented cylindrical pores. If made of semicrystalline material the resulting objects typically display a strong crystal orientation. Using polyvinylidenfluoride (PVDF) as a model system we investigated the crystallization and texture of polymers under 2D-confinement by DSC and X-ray diffraction. The crystal orientation in the nanorods and nanotubes can be explained by a model based on the kinetics of crystallization under conditions of confinement. Two clear limiting cases can be distinguished: If the nanorods are connected to a bulk-like surface film crystallization starts at low supercooling from a few heterogeneous nuclei in the surface film. Crystallization of the rods proceeds along their axes dominantly with the direction of fastest growth aligned parallel to the axes. In PVDF this results in a strong orientation of the (020)-plane perpendicular to the axes of the rods. The degree of orientation depends on the diameter of the rod or tube. A very different texture is found for isolated 1D-nanoobjects, i.e. in samples for which the surface film has been removed before crystallization. In this case in every rod a new nucleus has to form which can only be achieved by massive homogeneous nucleation at high supercooling. Due to the large number of randomly oriented nuclei initiating crystal growth, several growth faces contribute and the unique selection of one growth direction is lost. The ability to control the crystalline texture of 1D polymeric nanostructures is of potential interest for functional semicrystalline polymers, whose properties typically depend on crystal order and orientation.