
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
STRUCTURAL PROPERTIES OF POLY(ETHER) MACROMONOMER BASED HYDROGELS
P.J. LUTZ
Institut C.Sadron, UPR22, CNRS, F-67083 Strasbourg, e-mail: lutz / ics.u-strasbg.frThe major part of the present work discusses the properties of a series of hydrogels obtained by free radical polymerization (or by ATRP in some cases) of a,w-methacryloyloxy poly(ethylene oxide) (PEO) or poly(1,3-dioxolane) (PDXL) macromonomers.(1,2) The influence of several parameters such as macromonomer molar mass, solvent, and crosslinking concentration on the volume degree of equilibrium swelling, the uniaxial compression modulus and the thermodynamic interaction parameter was investigated systematically. The mechanical properties of hydrogels obtained in water are better than those of hydrogels obtained in an organic solvent. The influence of the presence of a comonomer on the mechanical properties of PEO or PDXL co-MMA networks was also studied.(3) Since poly(ether) macromonomers behave typically as amphiphiles, the rates of copolymerizations with hydrophobic comonomers such as MMA (methyl methacrylate) styrene, or butyl methacrylate are much higher than those of similar reactions carried in organic solvents. Hydrogels whose elastic chains are constituted of a short central PDXL block surrounded by two hydrophilic PEO blocks were prepared according to the same strategy.(4) PDXL is known to be sensitive to acidic degradation due to the presence of acetal groups. Therefore, once placed in acidic media (in water or in an organic solvent) network degradation should occur. The evolution, versus immersion time, of the number of elastically effective chains was followed by uniaxial compression modulus measurements and by solid-state 1H NMR spectroscopy. The solid-state properties of the different PEO or PDXL conetworks were examined by DSC, and compared to those of linear PEO or PDXL chains and to those of homopolymeric PEO or PDXL networks.
Such (PEO) macromonomer based hydrogels, accessible in water (or in physiological medium) are well-suited as a membrane for an artificial pancreas (1) and provide an interesting scaffold for cell adhesion and three-dimensional space for cell proliferation.
1) B. Schmitt, et al. Macromol. Biosci., 2002, 93, 341
2) K. S. Naraghi, P.J. Lutz et al. Polym. Int., 2002, 51, 912
3) N. Sahli, M. Belbachir, P. J. Lutz, Macromol. Chem. Physic., 2005 in press
4) K. Naraghi, B. Meurer, P. J. Lutz, Macromol. Rapid. Commun., 2005, 26, 537
EFFECTS OF ELECTROPHORETIC ION MIGRATION ON THE STRUCTURE OF AGAROSE GELS
R. BANSILa, A. MICHELMAN-RIBEIROa,b, R. NOSSALb
aPhysics Dept., Boston University, Boston, MA02215, USA (rb / bu.edu) bNational Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland 20892, USAThe influence of migrating ions on the microscopic structure of agarose gels subjected to an external electric field is not well understood. To address this issue we have performed 2-dimensional small angle light scattering (2D SALS) measurements on agarose gels in which ions migrate under the influence of E-fields ranging from 2-20 V/cm. We observe a macroscopic shrinking of the gel and anisotropic light scattering patterns indicative of domains oriented perpendicular to the E-field (See Figure below). Optical microscope imaging revealed anisotropic domains on the same length scale, also aligned perpendicular to the field. Video microscopy of gels doped with pH indicator dye show that ions migrating from both ends of the gel produce pH changes which are correlated with macroscopic shrinking and the anisotropic light scattering patterns. Profiles of pH variation across the gel, measured by video photography, reveal that the anisotropic patterns appear when the H+ and OH- ions migrating in opposite directions meet. Results of varying the valency and concentration of the ion will be presented. Calculations of pH profiles using a model based on electro-diffusion reproduce several features of measured pH profiles, including the E-field dependence of the time at which the oppositely charged fronts meet. Our observation of ion migration induced orientation and macroscopic deformation have implications for applications such as electrophoresis and the development of E-field responsive gels.
Figure Caption: An agarose gel (3% w/v) is deformed (right)
under the E-field applied in the direction indicated. The 2D SALS
pattern (left) shows pronounced horizontal lobes.
HYPERBRANCHED POLYMERS AS REACTIVE MATERIAL IN FILM AND COATING APPLICATION
B. VOIT, D. SCHMALJOHANN1, M. ABDELREHIM1, M. SANGERMANO2, A. DI GIANNI, E. PAVLOVA1,3, K. DUSEK3
1Leibniz Institute of Polymer Research Dresden e.V. (IPF);Hyperbranched polymers (HBP) have as dendritic macromolecules peculiar and often unique properties. They are characterized by a highly branched backbone, which give access to a large number of reactive groups. Their structure give them excellent flow and processing properties, which have attracted great attention. HBP with acrylate, vinyl ether, allyl ether or epoxy functions were studied as multifunctional crosslinker in coatings and in thermosets, using thermal as well as UV curing methods. Furthermore, the high density of functional groups in a confined environment renders those functional polymers also suitable as thin organic layers for sensor application.
We would like to report on the use of aromatic-aliphatic hyperbranched polyesters in UV curable coatings as well as vinylether functionalized crosslinkers1 as also as multifunctional transfer agents (phenolic end functions).2 The OH functionalized polyesters and hyperbranched polyols based on a poly(urea urethane) structures were also applied in thermally cured urethane coatings.
Hyperbranched polyesters based on bis-(4'-hydroxyphenyl)pentanoic acid as AB2 monomer were prepared in a typical melt polycondensation reaction. We modified the hyperbranched polyester with vinyl ether and oxetane functional groups for an application in UV curable coatings using cationic photoinitiators. Upon UV irradiation a very fast and nearly complete reaction of the vinyl groups was observed and the coatings showed sufficient mechanical stability in coating tests. However, without a diluent, only incomplete reaction of the vinyl groups was observed due to very fast reduction in mobility of the reactive groups in the highly crosslinked layer. Recently, the same hyperbranched polyester was also used directly without further modification in UV curable epoxy systems. It is known that OH containing molecules can act as transfer agents in the photoinitiated reaction. This fact could also be used to incorporate fluoromodified hb polyesters into a coating to yield low surface energy coatings.
The hyperbranched polyester was furthermore applied as polyol in classical thermally cured polyurethane crosslinking systems using 1.6-diisocyanatohexane (HDI) and its trimer (Desmodur N3300) as isocyanates. If polyfunctional components are involved resulting in formation of crosslinked polyurethanes, the kinetics becomes more complicated. The aromatic OH group involved in our case is considerably slower than that of aliphatic hydroxy group. Coating studies nevertheless showed that the hyperbranched polymer is a suitable OH-functional precursor for elevated temperature curable two-component high-solids polyurethane coatings for the application range up to 80-100 °C. The surfaces of films are smooth and relatively polar.
As a different polyol system, hyperbranched poly(urea urethane) polymers with aliphatic OH end groups prepared by a AA*+B*B2 approach have been applied in thermally cured systems also using 1,6-diisocyanatohexane (HDI) and its trimer as reactive monomers.3,4 Stable, highly crosslinked free standing films were obtained which showed good mechanical properties. Whereas, the product based on aromatic HBP is strong and relatively brittle, the aliphatic HBP led to a softer and more elastic network.
1) Schmaljohann, D.; Voit, B.; Jansen, J.F.G.A.; Hendriks, P.; Loontjens, J.A. Macromol. Mater. Eng. 2000, 275, 31-41. 2.) Sangermano, M.; Priola, A.; Malucelli, G.; Bongiovanni, R.; Quaglia, A.; Voit, B.; Ziemer, A. Macromol. Mat. & Eng.289, 442-446 (2004). M. Sangermano, A. Di Gianni, R. Bongiovanni, A. Priola, B. Voit, D. Pospiech, D. Appelhans, submitted to Macromol. Mater. Eng.(2005); 3.) Abdelrehim, M.; Komber, H.; Voit, B.; Langenwalter, J.; Bruchmann, B. J. Polym. Sci. Part A : Polym. Chem. 42, 3062-3081 (2004). 4.) Bruchmann, B. Phaenomen Farbe 2003, 2, 19.
SYNTHESIS AND PROPERTIES OF NANOCOMPOSITE AND PHOTOPOLYMER NETWORKS
C. DECKER
Département de Photochimie Générale (CNRS-UMR 7525) - Université de Haute Alsace - Ecole Nationale Supérieure de Chimie de Mulhouse - 3 rue Werner - 68200 Mulhouse (France) - C.Decker / uha.frHighly crosslinked polymers have been produced within seconds by photoinitiated polymerization of multifunctional monomers and oligomers. These ultrafast reactions were followed in situ by real-time infrared spectroscopy, a technique that records directly conversion versus time curves. The influence of chemical and physical factors on the polymerization kinetics has thus been quantitatively assessed for different types of resins (acrylate, epoxide, vinyl ether, thiol/polyene). Interpenetrating polymer networks have been readily produced by UV-irradiation of mixtures of multifunctional monomers polymerizing by different mechanisms, e.g. acrylates and epoxides (radical and cationic-type, respectively). The viscoelastic properties of the photopolymer networks have been finely tuned, through the chemical structure and molecular weight of the functionalized oligomers.
Such highly crosslinked polymers show an outstanding resistance to organic solvents and chemicals, and proved very effective as protective coatings to improve the surface properties and weathering resistance of polymer materials. The same technology was successfully used to produce nanocomposite materials, by a short UV exposure of a liquid resin containing mineral nanoparticles (colloidal silica or 1 nm thick silicate platelets). Exfoliation of the organoclay crystalline structure was demonstrated by X-ray diffraction spectroscopy and transmission electron microscopy. The in situ generated nanoparticles, which may assemble to form a skeleton-like structure, are considered to be responsible for the enhanced mechanical and gas-barrier properties. This novel method of synthesis of nanocomposite polymer networks presents the distinct advantages associated with the UV-curing technology, namely, a solvent-free formulation undergoing ultrafast polymerization at ambient temperature. Such performance, together with a cost-effective processing and enhanced properties, are key factors for the success of these newly developed photopolymer nanocomposites.
C.Decker, Macromol.Rapid.Comm. 23, 1067 (2002)
C.Decker, C. Bianchi, S. Jönsson, Polymer, 45, 5803 (2004)
S. Benfarhi, C.Decker, L.Keller, K.Zahouily, Europ.Polym.J. 40, 493 (2004)
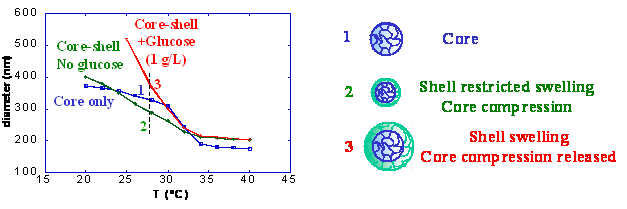
GLUCOSE-RESPONSIVE CORE-SHELL MICROGELS
V. RAVAINE, V. LAPEYRE, S. CHEVREUX, F. KORDASS
Laboratoire d'Analyse Chimique par Reconnaissance Moléculaire, E.N.S.C.P.B., 16 Av. Pey Berland, 33607 Pessac, France (v.ravaine / enscpb.fr ; http://www.enscpb.fr/lacrem)With the aim of developing new functionalized materials for sensors and drug delivery systems, we have synthesized a series of submicrometer-sized glucose-responsive microgels. Monodispersed thermosensitive microgels have been obtained by precipitation copolymerization of (N-isopropylacrylamide) (NIPAM) and different ratio of acrylamidophenylboronic acid (AAPBA). Their swelling behavior in various conditions has been investigated using dynamic light scattering. We first show that increasing the AAPBA concentration strongly decreases the swelling ratio of the microgels in water (pH=8.5) due to an increasing hydrophobicity, but does not affect the temperature of the volume phase transition. The addition of glucose induces the swelling of all microgels, even when the ionic strength of the solution is equal to 150 mM, proving the ability of these systems to potentially act as biological sensors.
Taking advantage of these results, we have synthesized core-shell particles constituted with a pNIPAM core and a p-NIPAM-co-AAPBA shell. We show that the core can be restricted from swelling due to the relative hydrophobicity of the shell. This compression vanishes upon addition of glucose (Figure 1), which opens new opportunities to design stimulating drug delivery systems.
 |
| Figure 1 |
DEVELOPMENT OF FLEXIBLE NETWORK POLYMERS CONSISTING OF OLIGOMERIC PRIMARY POLYMER CHAINS ORIGINATED FROM MULTIALLYL CROSSLINKING POLYMERIZATION
A. MATSUMOTO, M. DOURA, T. KIGUCHI, H. ITO, H. AOTA
Department of Applied Chemistry, Faculty of Engineering and HRC, Kansai University, Suita, Osaka 564-8680, Japan (amatsu / ipcku.kansai-u.ac.jp)It is well known that allyl monomers polymerize only with difficulty and yield polymers having low molecular weights, i.e., oligomers. Inevitably, free-radical multiallyl crosslinking polymerization1 provides network polymers consisting of oligomeric primary polymer chains, i.e., having abundant dangling chains. This was extended to the preparation of novel flexible amphiphilic network polymers (I) consisting of short primary polymer chains and long crosslink units with opposite polarities,2 simultaneous interpenetrating networks (II) consisting of both polyurethane (PU) and polymethacrylate (PM) networks with oligomeric primary polymer chains,3 and network polymers (III) consisting of centipede-type primary polymer chains.4
Thus, the solution copolymerizations of benzyl methacrylate with tricosaethylene glycol dimethacrylate in the presence of lauryl mercaptan yielded I consisting of nonpolar, short primary polymer chains and polar, long crosslink units. The opposite type of I was prepared by the copolymerization of 2-hydroxyethyl methacrylate, a polar monomer having a hydroxyl group, with heneicosapropylene glycol dimethacrylate, a nonpolar monomer having a poly(oxypropylene) unit. The equimolar polyaddition crosslinking reaction of poly(methyl methacrylate-co-2-methacryloyloxyethyl isocyanate) with tri(oxytetramethylene) glycol, leading to PU networks, and the free-radical crosslinking copolymerization of methyl methacrylate with tri(oxytetramethylene) dimethacrylate in the presence of CBr4, leading to PM networks, were progressed simultaneously, providing II formed via the topological crosslink between PU and PM network structures. The post-copolymerizations of oligomeric allyl methacrylate/alkyl methacrylate precopolymers, having different amounts of pendant allyl groups and different molecular weights, with allyl benzoate/vinyl benzoate monomer mixtures were conducted to give III.
1) A. Matsumoto, Adv. Polym. Sci., 123, 41 (1995); Prog. Polym. Sci., 26, 189 (2001). 2) M. Doura et al., Macromolecules, 36, 8477 (2003); J. Polym. Sci.: Part A: Polym. Chem., 42, 2192 (2004). 3) T. Kiguchi et al., J. Appl. Polym. Sci., 94, 1198 (2004); Macromolecules, 37, 8249 (2004). 4) A. Matsumoto et al., Polym Prepr. Jpn., 53, 394 (2004).
THREE DIFFERENT TYPES OF POLYMETHACRYLATE MODEL NETWORKS:
SYNTHESIS, CHARACTERIZATION AND MODELING
C.S. PATRICKIOS, M. VAMVAKAKI, A.I. TRIFTARIDOU, E. THEMISTOU, T.K. GEORGIOU, D. KAFOURIS, N. HADJIANTONIOU, M. KARBARZ
Department of Chemistry, University of Cyprus, POB 20537, 1678 Nicosia, Cyprus (costasp / ucy.ac.cy, http://www.ucy.ac.cy/~chemweb/ )Group transfer polymerization (GTP) chemistry was employed for the preparation of polymethacrylate networks of controlled structure (model networks) of three different types: (a) regular model networks, in which all polymer chains are linked at their ends, leaving, in theory, no free chain ends, (b) cross-linked star polymer model networks, in which star polymers are interlinked via half of their chains, letting the other half free (dangling), and (c) shell-cross-linked polymer model networks, in which the outer body of the network is decorated by polymeric arms (dangling chains). The linear and star precursors to the networks are characterized in terms of their molecular weight and composition using standard techniques such as gel permeation chromatography (GPC), dynamic and static light scattering and nuclear magnetic resonance (NMR) spectroscopy. Combination of hydrophilic and hydrophobic monomers within the networks leads to an amphiphilic character and a trend for microphase separation in water, which is investigated by swelling measurements, small-angle neutron scattering (SANS) experiments, and thermodynamic calculations. Introduction of a labile cross-linker within the networks results in degradable networks, whose stability is studied by GPC and NMR.
EFFECT OF MONOMER STRUCTURE AND IONIC COMONOMER COMPOSITION ON THE COIL-TO-GLOBULE TRANSITION IN HYDROGELS
V.R. Tirumalaa, J. Ilavskýa, M. Ilavskýb,c
aAdvanced Photon Source, Argonne National Laboratories, Argonne, IL 60439, USA bFaculty of Mathematics and Physics, Charles University, 180 00 Prague 8 , Czech Republic cInstitute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, 162 06 Prague 6, Czech Republic (vijay / aps.anl.gov. ilavsky / aps.anl.gov, ilavsky / kmf.troja.mff.cuni.cz)The coil-to-globule transition at lower critical solution temperature was studied using ultra-small-angle x-ray scattering in poly (N-alkylacrylamides) and poly (N-isopropylmethacrylamide). The collapse transition was found to be discontinuous in N-isopropylacrylamide (IPAAm) and N-isopropylmetha- crylamide (IPMAm) gels, while it was continuous in N,N-diethylacrylamide (DEAAm) gels. Addition of negatively charged sodium methacrylamide comonomer (xMna = 0.025 and 0.05) disrupted the chain collapse in N-alkylacrylamides but not in IPMAm gels.
We believe that the coil-to-globule transition in all three systems occurs due to hydrogen-bonding constraints imposed on thermal fluctuations. The collapsed state in IPAAm and IPMAm is preferred due to both increased entropy and decreased enthalpy while it is energetically unfavorable in DEAAm due to unpaired oxygen. The latter, therefore, undergoes continuous second-order transition. Addition of ionic comonomer introduces non-directional bonds to polymer backbone and thus decreases the hydrogen bonding constraints on thermal fluctuations.
A collapsed state for ionic copolymers is then not necessary although gels can still undergo macroscopic collapse by interchain hydrogen bonding across junction points. The presence of a-methyl group in IPMAm also decreases bonding constrains on thermal fluctuations (higher entropy) and charges would affect chain collapse in IPMAm only if the gain in conformational entropy due to the latter is greater than that from a-methyl groups.
Work at the Advanced Photon Source was supported by the Department of Energy, Office of Science, BES, under contract No. W-31-109-ENG-38 and Ministry of Education of the Czech Republic (grant MSM 0021620835).
Electrochemical Studies On Efficiency Of Solute Release From Thermoresponsive Poly(N-Isopropylacrylamide) Gels
W. HYK, M. KARBARZ
Department of Chemistry, Warsaw University, Pasteura 1, PL-02-093 Warsaw, Poland (wojhyk / chem.uw.edu.pl)In recent years much attention has been paid to the phenomenon of volume phase transition of gels due to their potential applications in drug delivery systems, separation techniques, and construction of sensors. The knowledge of gels' transport properties and efficiency of both loading the gel with various probe molecules and their release due to the gel phase transition is essential for the optimization of the application mentioned above.
This work presents an electrochemical method that allows one the simultaneous determination of the diffusion coefficient and efficiency of release of probe species from thermoresponsive gels due to their volume phase transition. The diffusion coefficient is a fundamental measure of species mobility in any medium while the release efficiency is measured by the release coefficient, θ, defined by the ratio of the concentrations of the probe in the expelled solution and the swollen gel. The method combines steady-state voltammetry at microelectrodes and conductometry, and it is designed for charged redox species. This method might be modified for uncharged systems; in this case instead of the conductometric measurement an additional independent voltammetric experiment in the solution is required.
The approach has been examined for charged and uncharged ferrocene derivatives as probes in the poly(N-isopropylacrylamide) hydrogel. The differences in the θ parameter, determined for charged and uncharged probe systems, are explained in terms of changes in dehydration of polymeric chains during the gel phase transition.
STRUCTURE FORMATION IN ISOTACTIC POLY(METHACRYLIC ACID)
E. VAN DEN BOSCH, H. BERGHMANS
Molecular and Nanomaterials, Catholic University of Leuven, Celestijnenlaan 200F, 3001 Heverlee, Belgium (edith.vandenbosch / chem.kuleuven.ac.be; hugo.berghmans / chem.kuleuven.ac.be)Investigations of the solution behaviour of stereoisomers of polystyrene and poly(methyl methacrylate) have shown that they can form thermoreversible gels by a mechanism very similar to the one that is responsible for the gelation of solutions of biopolymers like carrageenans. These stereoregular vinyl polymers can also crystallize in the "classical" way by a chain folding mechanism that leads to lamellar crystals [1]. These facts suggest that the process of thermoreversible gelation is quite general for polymer solutions as long as the correct experimental conditions can be realized. In order to find further evidence for this point of view, the possibility of thermoreversible gelation of a synthetic, water-soluble polyelectrolyte, isotactic poly(methacrylic acid) (iPMAA), was investigated.
iPMAA is not soluble in water at degree of neutralization a = 0 and solutions can only be prepared in hydrogen bond breaking solvents like DMF and DMSO. Amorphous films prepared from these solutions will only swell to a certain extend when brought in contact with water. The very rapid formation of a supramolecular organization manifests itself in several experimental observations. The films turn birefringent and specific changes in the infrared spectrum and the WAXD pattern take place. Solutions of this polymer in water can only be prepared above a certain critical a. For 10% (w/w) iPMAA solutions with a mass average molar mass of 10 kg/mol and an isotactic triad content of 92% this acrit is equal to 0.28 on neutralization with NaOH. Under these conditions thermoreversible gelation sets in on cooling around 0°C. Melting of such a gel takes place at much higher temperature leading to an important degree of hysteresis. This thermal treatment also leads to changes in the infrared spectra similar to those observed on swelling the polymer in water at a = 0. They are ascribed to an intramolecular coil to helix transition [2]. Dynamic mechanical measurements show that the process of structure formation is strongly influenced by shear. Structure formation can also take place at a > 0.4. This manifests itself in an increase in viscosity of the sample, in gelation or in polymer precipitation, depending on the polymer concentration, temperature and mechanical treatment of the sample. Preliminary data seem to suggest a mechanism different from that encountered at lower a.
[1] De Rudder, J.; Berghmans, H.; De Schrijver, F.C.; Bosco, M.; Paoletti, S. Macromolecules 35 (2002) 9529. [2] van den Bosch, E.; Keil, Q.; Filipcsei, G.; Berghmans, H.; Reynaers, H. Macromolecules 37 (2004) 9673.
PREPARATION AND SURFACE PROPERTIES OF POLY(DIMETHYLSILOXANE)- BASED INORGANIC/ORGANIC HYBRID MATERIALS
Z. LI, J. C. M. BROKKEN-ZIJP, G. DE WITH
Laboratory for Materials and Interface Chemistry (z.li / tue.nl), Eindhoven University of Technology , P.O. Box 513, 5600 MB Eindhoven, The Netherlands Laboratory for Materials and Interface Chemistry Laboratory for Materials and Interface Chemistry Laboratory for Materials and Interface ChemistryPoly(dimethylsiloxane) (PDMS)-based hybrid materials were prepared by the sol-gel method on Si wafer, Al and polystyrene (PS) substrates. The reaction was monitored by attenuated total reflectance-infrared (ATR-IR) spectroscopy. The hybrid materials made have surfaces in contact with air and different substrates. These surfaces were characterized by using tapping mode atomic force microscopy (AFM), X-ray photo-electron spectroscopy (XPS), low-energy ion scattering (LEIS), contact angle (CA) analysis and Johnson-Kendall-Roberts (JKR) technique. The hybrid surfaces contacting with air and substrates appeared to have different structures whereas the bulk properties were the same. The former have a ~1-2 nm thick silica-free PDMS top layer; while in the latter cases, SiO2 particles are located beneath the outermost atom layer, and - OH groups at the surface of SiO2 can easily stretch out when the surfaces contact with polar groups.
OPTIMISED NETWORK STRUCTURES FOR HIGH PERFORMANCE COATING SYSTEMS
C. FLOSBACH
DuPont Performance Coatings, Maerkische Strasse 243, D-42281 WuppertalThe structure of the final network after cross-linking is the most important key attribute of a high performance coating. Mechanical properties like scratch resistance and elasticity as well as chemical properties like etch resistance can be optimised by a tailored network structure.
To understand the network structure and the parameters influencing the network formation is essential to develop new coatings with superior performance.
A defined polymer architecture is necessary to achieve a homogeneous network formation, leading to the desired properties.
Characterisation methods like DMA and nano-scratch are used to control the network formation.
AN INTERPLAY OF SMALL ANGLE NEUTRON SCATTERING AND RHEOLOGICAL RELAXATION DATA IN DEFORMED POLYMER MELTS
W. PYCKHOUT-HINTZEN1, A. BLANCHARD1, D. RICHTER1, E. STRAUBE2 , D.J. READ3
1Forschungszentrum Jülich, IFF, D-52425 Jülich, D 2Universität Halle, FB Physik, D-06099 Halle, D 3University of Leeds, Appl. Math., LS2 9JT Leeds, UK Email: w.pyckhout / fz-juelich.deThe short time retraction process as well long time behaviour, following a fast step-strain deformation in a blend of linear low Tg polyisoprenes has been re-investigated by means of time-resolved but quenched small angle neutron scattering experiments. The new experiments find for the first time clear evidence for the non-linear process from scattering as well as from elongational rheology in the same conditions and combine information from linear rheology. The newly-designed rheometer allows simultaneous mechanical analysis and neutron scattering. The scattering results were interpreted in terms of the tube model [1] and the localization approach in terms of Warner-Edwards [2,3] for a network-like structure factor. The present relaxation mechanisms were modelled in a quasi-static way [4] and compare well to a full dynamic theory [5]. The retraction, describing forced contour length equilibration and reptation through the loss of tube constraints at the chain ends were simulated by assuming the chain deformation to be of triblock nature, the ends being isotropic. Constraint-release processes are treated in the dynamic dilution picture. The agreement of the tube dilution exponent with literature and especially the comparison of experimentally derived dangling end fractions with the estimates from fitting the tube occupation distribution from linear shear G',G"-rheology data is excellent.
[1] M. Doi, S.F. Edwards, The Theory of Polymer Dynamics, Clarendon (1989)
[2] M. Warner, S. F. Edwards, J. Phys. A 11 (1978) 1649
[3] D. J. Read, Eur. Phys. J. B 12 (1999) 431
[4] A. Blanchard, PhD Thesis Münster (2004)
[5] R. S. Graham, A. E. Likhtman, T.C.B. McLeish, S. T. Millner, J. Rheo. 47 (2003) 171
HEAT CAPACITY OF POLYMER LIQUID CRYSTAL SUBJECTED TO EXTERNAL DEFORMATIONS
J. WALASEK
Department of Physics, Technical University of Radom, 26-600 Radom, Poland (janusz9 / interia.pl)The polymer liquid crystal system (PLC) confined to longitudinal PLC chains is considered. The PCL longitudinal chain consists of liquid crystalline hard rods and flexible polymer sequences, alternately connected.
As a result of the non-lattice theory of externally deformed PLCs 1, the system entropy is a function of the chain end-to-end distances, while they are determined by the deformation.
Using that entropy to calculation of the system heat capacity, it is recognized how the external deformation can change in values that capacity.
In addition, it is shown that a change of the system architecture characterized by the concentration of the liquid crystalline particles in PLC, length of hard rods, length of the chain flexible sequences and the number of hard rods per chain, can vary, considerable, the heat of capacity.
1. Witold Brostow and Janusz Walasek J. Chem. Phys. Vol. 121, 7, 3272-3281 (2004)
DEGRADABLE THIOL-ACRYLATE AND THIOL-ENE SYSTEMS: NETWORK CONTROL FOR TISSUE ENGINEERING APPLICATIONS
S.K. REDDY1, A.E. RYDHOLM1, K.S. ANSETH1,3, C.N. BOWMAN1,2
1Chemical and Biological Engineering, University of Colorado, Boulder, USA(reddys / colorado.edu, rydholm / colorado.edu, kristi.anseth / colorado.edu, bowmanc / buffmail.colorado.edu) 2 Restorative Dentistry, University of Colorado Health Sciences Center, Denver, USA 3Howard Hughes Medical Institute, University of Colorado, Boulder, CO, USADegradable photopolymers are a unique class of biomaterials that have the ability to be injected into the body as monomer, polymerized in vitro using UV or visible light, provide matrix for tissue regeneration, and slowly degrade into benign byproducts. To be most effective for tissue engineering applications, these synthetic materials should be able to mimic a variety of material mechanics and tissue regeneration rates. This work focuses on the unique photopolymerization kinetics and network control aspect of thiol-acrylate and thiol-ene systems to form materials with a wide range of gel mechanics and degradation kinetics.
Thiol-acrylatephotopolymer networks evolve through a mixed step and chain growth mechanism, providing facile control of kinetic chain length distribution (KCLD) via changes in monomer functionalities and stoichiometries. A model that translates thiol-acrylate polymerization kinetics into network parameters successfully predicts the KCLD of gel permeation chromatograms. This understanding of KCLD was utilized in a bulk degradation model based on probability and mean field kinetics to predict successfully experimental mass loss profiles in thiol-acrylate systems. For the step growth polymerized thiol-ene systems, a degradation model based on probability and expectation theory was developed to predict gel degradation kinetics. In both systems a wide range of degradation kinetics were obtained and the mass loss at network dissolution ranged from as low as 25% to as high as 90%. In summary, these novel systems provide a simple route to control and manipulate gel degradation kinetics, without requiring elaborate monomer synthesis.