1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
THE GEL POINT AND NETWORK FORMATION - THEORY AND EXPERIMENT
J.I. CAIL, R.F.T. STEPTO*
Polymer Science and Technology Group, School of Materials, The University of Manchester, Grosvenor Street, Manchester, M1 7HS, U.K. *robert.stepto / manchester.ac.ukThe gel point, the point of incipient network formation, is an important point in network-forming polymerisations. Beyond the gel point, the viscosity of a polymerising mixture becomes infinite, a liquid-to-solid transformation starts to occur with the solid (network) fraction increasing as the polymersiation proceeds. In the reactive processing of non-linear polymerisations, liquid flow is no longer possible past the gel point. Therefore, in the fabrication of network materials in situ using reactive processing, it is important to be able to predict the gel point ab initio, from the formulation and the reaction conditions. In addition, the structure and properties of the network material finally formed can be related to the gel point of a polymerisation. Hence, the gel point can also be used to deduce what the physical properties of the final material will be.
This contribution examines the ab initio predictions and correlations of experimental gel points and network moduli at complete reaction for a range of model polymerisation systems, polyesters, polyurethanes and poly(dimethyl siloxanes). The analytical Ahmad-Rolfes-Stepto theory is shown to be the best basis for predicting amount of intramolecular reaction and gel point from reactant structures (molar masses, chemical functionalities, chain flexibilities) and reaction conditions (dilution, proportions of reactants). Due to the lengthy computations needed and the uncertainties in defining the gelation criterion for a polymerisation of finite size, Monte-Carlo (MC) simulation is found not to be a viable approach. However, MC simulation is shown to be essential for characterising the structure and modulus of a network at complete reaction, for which the whole distribution of sizes of ring structure is needed. In addition, it is demonstrated that the gel point can be employed to characterise the propensity of a polymerisation for intramolecular reaction and, hence, to predict network modulus at complete reaction.
THEORY OF EMBEDDABLE RANDOM NETWORKS
B.E. EICHINGER
Department of Chemistry, University of Washington, Seattle, WA 98118-1700Random networks that obey Flory-Stockmayer conditions have an exponential growth in the number of cross-links and chains as one progresses out from the center of the structure. Structures with an exponential growth cannot be embedded the three space with physically acceptable densities. Cascade theory, and variations thereof, relate the growth in the number of network chains with the number of generations (lengths of paths) of chains that grow out from a given root point or parent cross-link. The incorporation of cyclics into this scheme helps to attenuate this exponential growth, but eventually the embedding problem returns as no fractional attenuation of the exponential growth can suppress it altogether.
The embedding of a network in R
(Euclidean space of dimensions, not necessarily an
integer) is the result of a pre-existing distribution of
cross-linkable units (vertices) and chains (edges). Given a
uniform but random distribution of cross-links in R
and a real network that is built up from these cross-links and
adjoining chains, a graph can be constructed that faithfully
represents the structure. This is done by assigning directions to
edges, such that an edge points from the vertex closer to the
center to the one that is farther from it. Each vertex is
characterized by its Euclidian distance from the origin, by its
functionality or total degree, and by its in-degree defined by
the directed edges that are incident on it. Let ![]() be the number of
vertices of functionality f, in-degree k, at a
distance r from the origin embedded in R.
It is not difficult to write down a generating function giving
the probability of finding an
be the number of
vertices of functionality f, in-degree k, at a
distance r from the origin embedded in R.
It is not difficult to write down a generating function giving
the probability of finding an ![]() , given the assumption of equal and
random reactivity. The construction provides a new method for
calculating network statistics, and allows alternative estimates
to be made for gel points and related statistics.
, given the assumption of equal and
random reactivity. The construction provides a new method for
calculating network statistics, and allows alternative estimates
to be made for gel points and related statistics.
HYPERBRANCHED POLYMERS - THEORY vs. EXPERIMENT
H. GALINA, J.B. LECHOWICZ, M. WALCZAK
Rzeszów University of Technology, Faculty of Chemistry, 35-959 Rzeszów, PolandOne of the methods of preparing hyperbranched polymers is the condensation polymerization involving AB2 (or ARB2) monomers where A and B stand for functional groups reacting with each other (R is an organic moiety). This type of hyperbranched polymerization usually yields polymers of extremely broad molecular size distribution, particularly at high conversion of the minority A groups.
The molecular size distribution can be reduced at the expense of the average size of molecules by introducing a proportion of core monomer Bf having f reactive groups B of the same type as has AB2 monomer (f = 1, 2, 3, …). Further methods of narrowing the molecular size distribution are (i) carrying out the polymerization process in a semi-batch manner, i.e. by introducing AB2 monomer to Bf monomer either very slowly or divided into several portions, and/or (ii) extracting unreacted monomer from the polymer product after reaching certain conversion.
We have developed a kinetic model capable of assessing quantitatively the effects of these two methods and started to verify the predictions with the system: 4,4-bis(4'-hydroxyphenyl)-pentanoic acid (AB2 monomer) and phenol (B1), resorcinol, bisphenol A or butyl glycol (B2), 1,1,1-trihydroxymethyl-propane (B3) or pentaerythritol (B4).
The polymerizations were carried out under mild conditions (room temperature condensation) with dicyclohexylcarbodiimide water remover and piridine/aromatic sulphonic acid complex catalyst. The average molecular weights of polymers were recorded on a GPC apparatus equipped with low-molecular-weight columns and triple detector (RI, Visc., RALS).
ON THE NATURE OF CROSSLINKS IN THERMOREVERSIBLE GELS
K. TE NIJENHUIS
Delft University of Technology, Faculty of Applied Science, Department of Polymer Materials and Polymer Engineering E: k.tenijenhuis / tnw.tudelft.nlIt is well-known that many polymers, synthetic and natural, form physical, thermoreversible aggregates in dilute solution, whereas in moderately concentrated solutions gels can be formed. Examples are poly(vinyl chloride), polyacrylontril, poly(vinyl alcohol), atactic polystyrene, mixtures of syndiotactic and isotactic poly(methyl methacrylate), liquid crystalline polymers, gelatin, agarose, carrageenans etc. These gels in general belong to the third type in the classification of Flory [1]: polymer networks formed through physical aggregation, predominantly disordered, but with regions of local order. Schematic views of the various kinds of thermoreversible gels are shown in the Figure [2]. In PVC, PVA, PAN and PE micellar crystallites are responsible for the gel formation (A), whereas helix formation (B) is the origin of the biopolymer gels of gelatin, agarose, carrageenan and gellan gum. Phase separation (C, D and E) causes network formation in solutions of atactic polystyrene and in solutions of ABA block copolymers in selective solvents, where the phase separated A-blocks are not soluble in. In these cases (C and D) the crosslinks do not consist of ordered regions (like in Flory's classification), but of relatively dense amorphous regions, unless the A-blocks are semi-crystalline (E). Gelation can also find its origin in complex formation (F): physical complexes in mixtures of isotactic and syndiotactic poly(methyl methacrylate) or chemical complexes in PVA/Congo red, PVA/Borate and partly hydrolysed Poly(acryl amide)/Cr3+. In liquid crystalline polymer solutions gelation will be caused by interaction between mesogenic groups in the main chain and/or in the side chain (G).
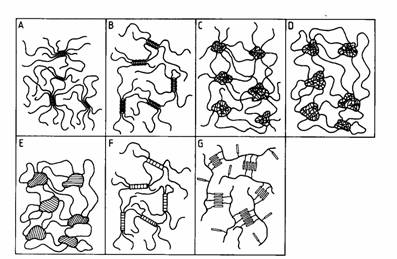 |
References
[1] P.J.Fory, Faraday Discussions of the Chemical Society, 57 (1974) 7-18
[2] K.te Nijenhuis, Adv.Polym.Sci.,130 (1997) 1-252
BIOPOLYMER GELATION- EXPONENTS AND CRITICAL EXPONENTS
S.B. ROSS-MURPHY
Molecular Biophysics Research Group, Department of Life Sciences, King's College London, Franklin-Wilkins Building, 150 Stamford Street, Waterloo, London SE1 9NH, UK (simon.ross-murphy / kcl.ac.uk)The gelation of biopolymer systems has been studied, at least, empirically for many years, but only more recently have the methods of macromolecular science been applied. Many ensuing studies have tended to concentrate on measuring power law exponents, and ignored the effects of the network structure.
However, by their very nature, biopolymer physical gels are more complicated than "simple" chemically cross-linked systems. Almost all form from aqueous solutions but some develop on cooling and some on heating. In turn some of these are thermoreversible, some are not.1, 2 This means that approaches designed, for example, for cross-linked melts have to be applied with caution. Consequently, while measuring exponents alone can give some valuable information, and some apparent "universalities" are seen, the details of pre-exponential factors can often prove more significant and useful.
Using these systems to establish the criticality of exponents has been attempted many times. In the authors' view, few, if any, of these studies have been useful, because measuring the properties of all systems in the critical region, when p/pc ~1, with pc the usual percolation threshold, is difficult for model cross-linking systems, and becomes more challenging still for this class of materials.
One consideration is that critical exponents are defined only when data are accessed within the critical region. This, in turn, is defined by criteria such as that of Ginzburg. Evaluation of the extent of the critical domain remains the realm of the theorist but a practical guide is that 10-2 <= (p/pc)-1 <= 10-1.3 It appears that very few published data satisfy this, albeit demanding, requirement. This, coupled with the limited range of viscoelastic linearity close to the gel point, would suggest it is very difficult to confirm that measured values are really "critical exponents".
1. Clark, A. H.; Ross-Murphy, S. B. Adv. Polym. Sci. 1987, 83, 57.
2. te Nijenhuis, K. Adv. Polym. Sci. 1997, 130, 1.
3. Stauffer, D.; Coniglio, A.; Adam, M. Adv. Polym. Sci. 1982, 44, 103.
POLY(IMIDE-AMIDE) - POLY(ETHYLENE GLYCOL) HYBRID NETWORKS: NANOSTRUCTURE, MOLECULAR DYNAMICS AND MEMBRANE PROPERTIES
V. A. BERSHTEINa, L.M. EGOROVAa, P.N. YAKUSHEVa, V. SINDELARb, P. SYSELb, T.E. SUKHANOVAc, I.P. DOBROVOLSKAYAc, A.I. GRIGORIEVc, S. KRIPOTOUd, P. PISSISd
aMaterials Dynamics Laboratory, Ioffe Physico-Technical Institute of RAS, 26 Politechnicheskaya str., 194021 St. Petersburg, Russia (vbersht / polmater.ioffe.ru) bDepartment of Polymers, Institute of Chemical Technology, 5 Technicka, 16628 Prague 6, Czech Republic cPolymer Morphology Laboratory, Institute of Macromolecular Compounds of RAS, 31, Bolshoy pr. V.O., 199004 St. Petersburg, Russia dDepartment of Physics, National Technical University of Athens, Zografou Campus, 15773 Athens, GreecePoly(imide-amide)s cross-linked with poly(ethylene glycol) were synthesized from poly(amic acid) based on pyromellitic dianhydride (PMDA) and 4,4'-oxydianiline (ODA), and PEG terminated with 4-methyl-1,3-phenylene diisocyanate. The PIA-PEG networks differed in PEG molecular weight (Mn=1000 or 3400 g/mol) and weight content (from 20 to 80%).Combined WAXD/SAXS/polarized microscopy/DSC/DRS/TSDC/creep rate spectroscopy (CRS) analysis of structure and molecular dynamics (at 130-570 K), as well as estimation of the membrane properties were performed for these hybrid networks.
The hybrids with PEG(1000) cross-links were amorphous, whereas different structural state, semi-crystalline, at 20-40 wt. % PIA, and mesomorphous or amorphous, at 50-80 wt. % PIA; was observed for PEG(3400) cross-links in the networks; PEG crystallinity decreased from 55 to 0% with increasing PIA content in a hybrid. Nanostructural heterogeneity was found in some of these hybrids, with two kinds of scattering domains, of Rg1»10 nm and Rg2»700 nm in size. The hybrids under study could be subdivided into two groups: (a) PEG-rich networks with a continuous PEG phase and a single glass transition I at Tg1DSC = 220-250 K, and (b) PI-rich networks with "suppressed" glass transition I and spatially isolated PEG domains but with the distinct glass transition II at Tg2DSC = 550-570 K (PIA domains). The discrete CR spectra were obtained over the temperature range from ~200 to 550 K, which indicated large heterogeneity of segmental dynamics in the hybrids studied, in particular the effects of "constrained" dynamics in PEG cross-links.
The membrane properties of the networks studied turned out in a good accordance with dynamic behavior of PEG cross-links. In contrast to the networks with "suppressed" PEG mobility and low permeability, the permeability coefficients of films with 60-80%PEG were higher, by 2 or 3 orders of magnitude, for organic vapors than those of air gases.
POLYCYANURATE NETWORKS MODIFIED BY FLEXIBLE CROSSLINKS
P. Pissisa, S. Kripotoua, E. Kontoub, A. Fainleibc, O. Grigoryevac, I. Beyc, V.A. Bershteind, V. Egorovd, P. Yakushevd, L. Davide
aDepartment of Physics, National Technical University of Athens, Zografou Campus, 157 80 Athens, Greece (ppissis / central.ntua.gr) bDepartment of Mechanics, National Technical University of Athens, Zografou Campus, 157 80 Athens, Greece cInstitute of Macromolecular Chemistry, National Academy of Sciences of Ukraine, 02160 Kiev, Ukraine dIoffe Physico-Technical Institute, Russian Academy of Sciences, 26 Polytechnicheskaya str., 194021 St Petersburg, Russia eUniversite Claude Bernard Lyon I, UMR 5627, LEMPB, 43 Bd du 11 Novembre 1918, 69622 Villeurbanne, FranceDensely cross-linked polycyanurate networks (PCN) exhibit high glass transition temperatures and high thermal stability, good mechanical properties, low dielectric constants and water uptakes, and high chemical resistance. Their major drawback with respect to applications in high performance technologies is high brittleness. Several attempts are currently being undertaken to improve this particular aspect. In this work, hybrid PCN - poly (tetramethylene glycol) (PTMG) networks were prepared and their structure/morphology, molecular dynamics and thermal/mechanical properties were investigated by several experimental techniques.
Several series of samples with varying molar mass and content of PTMG were prepared and studied. Hybridization was confirmed by FTIR spectroscopy and sol-gel analysis was used to determine the fraction of PTMG incorporated into the PCN network. Wide- and small-angle X-ray scattering (WAXS and SAXS, respectively) was used for morphological characterization. Various nanostructures were observed depending on PTMG molar mass and fraction.
Thermal transitions and molecular dynamics were investigated by differential scanning calorimetry (DSC), thermally stimulated depolarization currents (TSDC), creep rate spectroscopy (CRS) and broadband dielectric spectroscopy (DRS). CRS/DSC analysis revealed a complicated dynamic behavior explained in terms of a wide dispersion of glass transitions in the hybrid networks due to the presence of nanodomains with different degrees of rigid crosslinking. TSDC and DRS showed a broad α relaxation in the hybrids, associated with the main glass transition, which shifts to lower temperatures with increasing PTMG fraction, and two local relaxations, γ of PTMG and γ of PCN. Finally, stress-strain measurements provided information on the mechanical performance of the hybrids, which was correlated with structure and molecular dynamics.
MULTICOMPONENT POLYMER NETWORKS PRODUCED BY POLYMERIZATION-INDUCED PHASE SEPARATION
R.J.J. WILLIAMS, I.A. ZUCCHI, M.J. GALANTE
Institute of Materials Science and Technology, University of Mar del Plata and National Research Council, J. B. Justo 4302, 7600 Mar del Plata, Argentina (intema / fi.mdp.edu.ar)Polymerization-induced phase separation in systems of two different modifiers dissolved in thermoset precursors leads to a variety of morphologies and properties of the resulting multicomponent polymer networks. Depending on the mutual compatibility among the different components a single or a double phase separation process may take place. Particular examples showing both types of behaviours will be described.
Polystyrene (PS), poly(methyl methacrylate) (PMMA), and PS-b-PMMA, were used as modifiers of an epoxy-amine formulation. Both PS and PMMA were initially miscible in the monomers at 80 ºC. A solution containing 5 wt % of each one of both linear polymers exhibited a double phase separation process; first a PS-rich phase was segregated followed by a PMMA-rich phase at higher conversions, leading to a dispersion of individual PS and PMMA particles in the epoxy network. A solution of 10 wt% of the block copolymer in the epoxy-amine monomers led to a completely different morphology. PS blocks were not initially miscible and formed micelles stabilized by PMMA chains. At the time of PMMA phase separation coalescence of the micellar structure was produced leading to a thermoplastic phase that percolated the epoxy network. This produced a significant decrease in the yield stress necessary for toughening purposes.
Biphenyl (BP), polystyrene (PS) and several types of epoxy networks exhibit similar values of refractive indices above the melting point of BF. Thermally-reversible light scattering films (TRLS) were obtained in a single phase separation process during polymerization of formulations containing the three components. Morphologies consisted of droplets of BF/PS solutions dispersed in an epoxy matrix plasticized by a fraction of residual BF. Crystallization of BF was confined to the interior of droplets and changed the optical state of the multicomponent blend from a transparent to an opaque state. These TRLS films exhibited much better optical and mechanical properties than binary films synthesized with BF and the epoxy precursors. Replacement of BP by an organic liquid crystal (EBBA) enabled to reduce the undercooling necessary to produce the ordered phase.
BISMALEIMIDE-DICYANATE-RESINS (BT-RESINS) - NEW INSIGHTS IN CATALYSIS, MECHANISM, AND RECYCLING
W. MORMANN, S. FLEISCHMANN
Universität Siegen, Laboratorium f. Makromolekulare ChemieBismaleimide triazine resins have been marketed since a long time; they are known for their excellent high-temperature stability, insulation resistance and dielectric properties. Two curing mechanism have been put forward; the older assuming formation of a cyclotrimer from one maleimide and two cyanate groups, a newer postulates formation of interpenetrating maleimide and cyanurate networks.
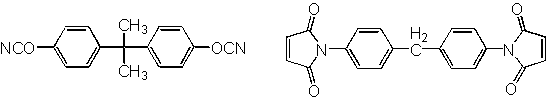
We studied the curing mechanism of cyanate/maleimide systems with phenylcyanate and phenylmaleimide as model compounds in the presence of different amine catalysts. At 100 °C phenylcyanate did not react in the presence of DABCO. After addition of catalytic amounts of phenyl maleimide fast reaction of phenyl cyanate occurred. Reaction products were identified by HPLC/MS or with authentic samples. Cyclotrimers and linear oligomers of phenylcyanate as well as cyclic and linear oligomers of phenyl maleimide are formed under these conditions.
Mixed oligomers are in agreement with HPLC/MS results.
Chemical recycling of polycyanurates and BT-resins by ammonolysis will also be reported.
Unusual case of network polymer formation IN anionic polymerization of monofunctional vinyl monomer*
B. Rozenberg
Department of Polymers and Composite Materials, Institute of Problems of Chemical Physics Russian Academy of Sciences, Semenov av. 1, Chernogolovka Moscow region, 142432, Russia, rozen / icp.ac.ruHydroxyalkyl (meth)acrylates are monofunctional vinyl monomers. They form usually linear polymers with isomerized polyester-ether backbone [1-6] under the conditions of anionic polymerization. It was surprising to obtain network polymer at 2-hydrohyethyl-methacrylate (HEMA) polymerization under the action of potassium [2-6]. Using isothermal calorimetry, IR,1H and 13C NMR spectroscopy, exclusion and liquid chromatography under critical conditions, and some other experimental and theoretical approaches the kinetic features and mechanism of HEMA polymerization as well as structure of polymer formed were elucidated. The lecture is devoted to discussion of the mechanism, which is responsible for such unusual behavior of HEMA anionic polymerization.
References
Rozenberg B.A., Bogdanova L.M., et al., Polym. Sci., Ser. A. 45, No.1, 5 (2003).
Rozenberg B.A.,Int. J. Plast. Techn., 6, No 1, 17 (2003).
Estrina G.A.,Komarov B.A., et. al., Polym. Sci., Ser. A,. 46, No 2, 97 (2004).
Rozenberg B.A., Boiko G.N., et. al., Polym. Sci., Ser. A. 46, No.3, 1429 (2004).
Rozenberg B.A., Designed Monomers and Polymers, 7, No 1-2, 135 (2004).
Rozenberg B.A., Irzhak V.I., et al., Polym. Sci., Ser. A. 47, No.3, 226 (2005).
*This work was supported by the Russian Foundation for Basic Research (Project 04-03-32674a), Division of Chemistry and Material Sciences Russian Academy of Sciences (Project 04-12-131), and the International Science and Technology Center (Project 02-1918.)
CELLS, GELS AND THE ENGINES OF LIFE: A POLYMER GEL APPROACH TO CELL FUNCTION
G.H. POLLACK, J.M. ZHENG
Department of Bioengineering, University of Washington, Seattle WA 98195The cell is a gel. However, the cell's gel-like character has not been broadly exploited for understanding cell function, except for a few notable examples. These include the action potential (Tasaki), secretion (Verdugo, Fernandez), muscle contraction (Pollack), and protein folding (Urry), where abundant evidence shows that the central player in each of these phenomena is the polymer gel phase transition. The thesis advanced here is that the polymer gel phase transition transcends these examples, and is in fact a generic mechanism for cell function. Evidence for this thesis is offered in a recent book, Cells, Gels and the Engines of Life (www.ebnerandsons.com).
One of the proposed central features of the generic phase transition mechanism is the state of water. Ample evidence shows that water inside the cell is not bulk, but is largely structured. This follows from the extreme crowding of cellular macromolecules, leaving narrow intermolecular spaces that make cell water largely interfacial.
To follow up on the issue of water structuring, experiments have been carried out on the interfacial region adjacent to hydrophilic surfaces. We found that solutes were excluded from this region (Zheng and Pollack, Phys Rev. E. 68: 031408, 2003). The solute-exclusion zone extends typically 100 µm or more from the surface, a span well beyond expectation. Exclusion-inducing surfaces include those of various polymers, cells, hydrogels, and ion-exchange beads. Excluded entities range from 10-µm microspheres, to proteins, all the way down to dyes of molecular weight several hundred Daltons. Hence, the solute-exclusion phenomenon is both long range and general.
Current experiments address the mechanism underlying this surprising phenomenon. Results to date imply that water in the exclusion zone is physically different from water more distant from the surface. These results include NMR experiments showing shortened T2 values, infrared images showing reduced radiation, and potential measurements showing large negative potentials in this zone. On the basis of these results, we tentatively conclude that hydrophilic surfaces may impact water out to unexpectedly large distances.
PROPERTIES OF FISH PROTEIN - POLYMER HYBRID GELS
M. FURUKAWA1, K. KOJIO2, Y. SAKAMOTO1, Y. MINAMIDA2
1Department of Materials Science, Graduate school of Science and Technology, Nagasaki University, 1-14 Bunkyou-machi, Nagasaki, 852-8521 Japan, 2Department of Materials Science and Engineering, Faculty of Engineering, Nagasaki UniversityMuscles transform the chemical energy, including an external stimulus, into mechanical energy efficiently, resulting in the reversible contraction and relaxation. Myosin, which can change a chemical energy into "a motion" in the body, is the main component of muscle, and is the huge protein with the molecular weight of about 480,000 and a molecule length of 150 nm. All kinds of muscular myosin of animal also change structurally by heating (denaturation), and the hydrogel can be formed by heating with a salt. In this study, the bio-hybrid gels were prepared by fish protein gel with synthetic polymer and evaluated their properties.
| The muscular protein used
was extracted from fish meat. After washing and
centrifugal separation, the muscular protein was mixed
with 3wt% of NaCl and the mixture was grounded. The fish
protein gels were prepared by heating the mixtures at
various temperatures from 50 to 90 °C. The bio-hybrid
gels were prepared from fish protein gel and polyvinyl
alcohol. .Young's modulus, tensile strength and
elongation at break of the bio-hybrid gels increased with
an increasing PVA content. Mechanical properties of these
gels were measured under the electric stimulus. Figure 1
shows the bending angle of the fish protein gel and the
hybrid gels in various pH solutions under the electric
stimulus. The bending behavior of the hybrid gels was
similar behavior of the fish protein gels. These gels
showed the largest bending angle (20-300) and
responded load in pH=1.2. The other hybrid gels as a capsule were prepared by coating myofibril gel with biodegradable polyurethane. The behavior of the micro-capsule gels as a drug delivery gel will be also discussed. |
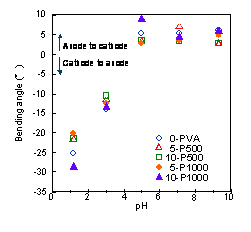 |
| Figure 1: Bending angle of fish protein gel and hybrid gels in various pH solutions under the electric stimulus |
NATURAL RUBBER NETWORKS AS A SMART NANOCOMPOSITE BY REVERSIBLE CRYSTALLIZATION UPON STRETCHING AND RETRACTION
S. KOHJIYA
Kyoto University, Institute for Chemical Research, Gokasho, Uji, Kyoto 611-0011, Japan (e-mail: kohjshin / scl.kyoto-u.ac.jp)Powerful X-ray sources are available for scattering or diffraction studies at a synchrotron facility. They enable us to carry out dynamic i.e. time-resolved measurements of wide-angle X-ray diffraction on natural rubber (NR) during its stretching and retraction cycle. Crosslinked NRs were subject to the measurements together with usual evaluations e.g. tensile and dynamic mechanical measurements. The most surprising feature is that the majority of rubber network chains remains to be unoriented amorphous (possibly Gaussian) state even under high elongations e.g. 600-% elongation. The amount of crystallized and oriented amorphous combined was at most 40 %. In other words, 60-% network rubber chains may remain to be without any stress even at 600-% elongation! Our experiment findings strongly suggest that the Affine deformation may not be the case for the deformation behavior of crosslinked NRs, although it has been considered to be an excellent approximation for overall description of rubber elasticity. The retraction of the stretched networks recovered the initial amorphous state, which implies the reversible crystallization-melting upon stretching-retraction cycle. The strain-induced crystallites did contribute to the increase of modulus. In other words, NR networks are smart enough to reinforce by themselves when necessary (under a high strain larger than ca. 300-% strain). The crystallites formed upon elongation were the size of nanometers, which were of use for self-reinforcement. Also, when they were compounded with reinforcing carbon black (HS-HAF), both carbon black and the crystallites formed at higher elongations were found to be contributing to their mechanical properties. NR networks are smart enough to accommodate the reinforcer while maintaining the crystalizability under a high strain.
NETWORK STRUCTURES AND STRESS RELAXATION IN POLYMERS FORMED FROM THIOL - ENE POLYMERIZATIONS
S.K. REDDYa, T.F. SCOTTa, C.N. BOWMANab
a Chemical and Biological Engineering, University of Colorado, Boulder, USA (reddys / colorado.edu, timothy.scott / colorado.edu, bowmanc / buffmail.colorado.edu) b Restorative Dentistry, University of Colorado Health Sciences Center, Denver, USAFree radical photopolymerizations, which rapidly transform liquid monomers to crosslinked polymers, have been a rapidly growing industry for many years now. While this exciting field is currently dominated by (meth)acrylic formulations, thiol-ene photopolymerizations are increasingly becoming an attractive alternative due to their superior cure kinetics and controlled network evolution aspects. In thiol-ene polymerizations, the network evolution occurs through a pure step growth framework when the ene monomer is non-homopolymerizable and through a mixed step-chain growth framework when the ene monomer is homopolymerizable. Due to their unique polymerization behavior, the formed thiol-ene polymers exhibit attractive properties that include delayed gelation, low polymerization stresses, and homogeneous network evolution. Further, the network control aspects of these systems in association with the network evolution model we developed enabled us to form systems with two orders of magnitude variance in modulus and homogenous polymers with a wide range of glass transiton temperatures.
The incorporation of addition-fragmentation chain transfer groups in the thiol-ene polymer backbone allows for stress and/or strain relaxation to occur in these networks. Such groups may be introduced by either copolymerizing the thiol-ene system with allyl sulfide ring opening monomers (whose propagation mechanism via a sulfur centred radical allows them to be added to thiol-ene systems without altering the stoichiometry of the system) or alternatively through the incorporation of the functional group in the backbone of one or both of the thiol-ene monomers. The network structure resulting from the step-growth nature of the polymerization makes these networks particularly suited to this stress relaxation method. Such stress relaxation processes lead to dramatic photoinduced plasticity, actuation, and equilibrium shape changes.
INTERFACIAL REGIONS IN PHASE-SEPARATED INTERPENETRATING NETWORKS
YU.S. LIPATOV
Department of Physical Chemistry of Polymers, Institute of Macromolecular Chemistry, National Academy of Sciences of Ukraine, Kiev 02160, Ukraine (lipatov / imchem.kiev.ua)Interpenetrating polymer networks (IPN) are, as a rule, two-phase systems due to the phase separation proceeding during the reactions of their formation. The characteristic features of the processes of IPN formation have been studied experimentally for various full and semi-IPNs. It was established that the main role in the process belongs to the superposition of the chemical kinetics and of physical kinetics of phase separation, its driving force being the appearance of the thermodynamic incompatibility of two components at a definite conversion degree. Phase separation in IPNs obeys to the spinodal mechanism. Due to the simultaneous proceeding of two processes, phase separation proceeds in the nonequilibrium conditions being dependent on the reaction kinetics. As a result of the high viscosity of the reaction media and diffusion limitations the phase separation stays uncompleted. As a result there appears an interfacial region between two evolved phases. The segregation degree of the system into two phases, the composition of phases and fraction of interfacial region depend on the kinetic conditions of reactions. The segregation degree and the fraction of interfacial region may serve as a measure of thermodynamic nonequilibrium of the system. These characteristics were determined from the measurements of the viscoelastic properties and thermophysical properties. The structures developing at the initial stages of reaction and phase separation are fixed by reaching the gel point. The completion of both reactions proceed in two evolved phases.
Interfacial region preserves the level of molecular mixing of components before the onset of phase separation (" frozen" compatibility). The higher is the fraction of an interfacial region, the less is the segregation degree and the higher is the deviation of the system from the state of thermodynamic equilibrium. The fraction of interfacial region may be regulated by the change of the kinetic conditions of reaction and by introducing compatibilizing agents and fillers into the starting reaction system.
LATENT CURING TENDENCIES OF CYANATES, EPOXIES AND VINYL ETHERS WITH A FERROCENIUM PHOTOCURING SYSTEM
W.D. COOK
School of Physics and Materials Engineering, Monash University, Clayton, Victoria, Australia (wayne.cook / spme.monash.edu.au)The most widespread method of photopolymerization utilizes free-radical initiation but other initiation methods, in particular cationic photopolymerization, are becoming more common. In contrast to free radical cure, where the polymerization usually terminates within seconds of cessation of irradiation, it has been reported that cationic polymerization can continue for hours after cessation of irradiation, suggesting that initiating or propagating species remain active for a considerable period. In this paper, we report on the photo-DSC and photo-rheometry of 1,1-bis(4-cyanatophenyl)ethane(BCYPHE), diglycidyl ether of bisphenol-A (DGEBA) and tri(ethylene glycol) divinyl ether (TEGDVE) using a 3M Visilux dental curing light (lmax » 470nm) and cyclopentadienyl fluorene iron hexafluorophosphate (CPDF) photointiator.
The cationic photoinitiation by a ferrocenium is believed to involve four steps - photolysis of the initiator, coordination of the initiator by the monomer, carbonium ion formation and cationic propagation. The photolysis and propagation are rapid but it is not known whether coordination or carbonium ion formation is slowest. Mossbauer spectroscopy shows that two FeII cyclopentadienyl species exist during the photopolymerization which is consistent with the presence of unchanged CPDF and the FeII cyclopentadienyl coordinated with the monomer ligands.
The photocuring behaviour of the cyanate ester after varying periods of irradiation reveals that a dark cure continues for more than 10 minutes and suggests that either the formation of active species or the propagation of cations continues for long periods. Even stronger evidence of dark cure is found for the cationic photopolymerization of DGEBA. The photopolymerization of TEGDVE shows even more extreme dark reaction behaviour - polymerization does not occur during steady illumination at room temperature nor of the same sample when heated in the dark to 70°C until an extended induction period has elapsed. The potential for using this system in adhesive applications was illustrated by irradiation of the divinyl ether at room temperature and then heated to 50°C in the dark - the material remained liquid for about 3 minutes before significant polymerization commenced leading to a rubbery material within another 2 minutes.