Postery
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33
ALTERNATIVNÍ METODY PŘÍPRAVY N-KARBOXYANHYDRIDŮ
α-AMINOKYSELIN
M. VAVŘÍKa, J. CZERNEKa
aOddělení bioanalogických a speciálních polymerů, Ústav
makromolekulární chemie AV ČR, Heyrovského náměstí 2, 162 06 Praha 6,
telefon +420-296809290, email {vavrik, czernek}/imc.cas.cz
V současnosti jsou intenzívně zkoumány biomateriály vytvářené
samoasociací bloků poly(α-aminokyselin) [1]. Příslušné
poly(α-aminokyseliny) jsou nejčastěji připravovány "živou" polymerací N-karboxyanhydridů
α-aminokyselin mechanizmem aktivovaného monomeru [2] nebo s využitím
komplexů přechodových kovů jako katalyzátorů [3]. Potřebné
N-karboxyanhydridy (NCA) jsou obvykle syntetizovány přímou
reakcí dané α-aminokyseliny s fosgenem nebo trifosgenem. Tyto metody
mají značné praktické nevýhody. Kromě práce s vysoce jedovatým fosgenem
je pro ně charakteristický vznik obtížně odstranitelných reaktivních
vedlejších produktů, které limitují možnosti přípravy NCA v čistotě
dostatečné pro řízenou polymerizaci. Zde se zabýváme alternativními
přístupy k syntéze NCA, konkrétně reakcí α-aminokyselin s di-tert-butyltrikarbonátem
(Schéma 1) a využitím chráněných α-aminokyselin k reakci s
trimethylchlorsilanem následovanou cyklizací oxalylchloridem (Schéma
2). Jde o modifikace syntéz popsaných v literatuře [4], resp. [5].
Di-tert-butyltrikarbonát reaguje s α-aminokyselinou v ultrazvuku za vzniku
N-karboxyanhydridu α-aminokyseliny. Výhodou metody je, že odpadními produkty jsou oxid uhličitý a tert-butanol,
jež lze bez potíží odstranit (nezreagovaná α-aminokyselina se
odfiltruje). Získaný NCA není většinou potřeba dále čistit. Na základě
hodnot reakčních výtěžků je zřejmé, že tato metoda je vhodná zejména
pro přípravu N-karboxyandridů L-alaninu a γ-benzyl-L-glutamátu.
Druhá metoda využívá reakce chráněné aminokyseliny (např. N-Boc-L-alaninu) s trimethylchlorsilanem za vzniku jejího trimethylsilylesteru 1 (Schéma 2). Následuje cyklizace 1
za použití oxalylchloridu. Výhodou tohoto postupu jsou mírné reakční
podmínky a také to, že doba přípravy včetně čistících procedur obvykle
netrvá více než pár hodin.
Popsané metody nám umožňují efektivní přípravu většího množství (desítky gramů)
N-karboxyanhydridů těch α-aminokyselin, které využijeme pro
syntézu blokových kopolymerů s cílem získat hydrogely s rheologickými
vlastnostmi vhodnými pro aplikace v tkáňovém inženýrství.

Schéma 1 Schéma 2
Literatura
1. Nowak, A. P.; Breedveld, V.; Pakstis, L.; Ozbas, B.; Pine, D. J.; Pochan, D.; Deming, T. J. Nature 2002, 417, 424.
2. Dvořák, M.; Rypáček, F. Chem. Listy 1995, 89, 423.
3. Deming, T. J. J. Polym. Sci. Part A: Polym. Chem. 2000, 38, 3011.
4. Nagai, A.; Sato, D.; Ishikawa, J.; Ochiai, B.; Kudo, H.; Endo, T. Macromolecules 2004, 37, 2332.
5. Mobashery, S.; Johnston, M. J. Org. Chem. 1985, 50, 2200.
SYNTÉZA BIODEGRADOVATELNÝCH BLOKOVÝCH KOPOLYMERŮ NESOUCÍCH BIOAKTIVNÍ PEPTIDOVÉ SEKVENCE
V. PROKS, L. MACHOVÁ, F. RYPÁČEK
Ústav makromolekulární chemie AV ČR, Heyrovského nám. 2. Praha 6, 162 06
(proks/imc.cas.cz)
Alifatické polyestery jsou pro svoje biodegradovatelné vlastnosti
často využívány jako materiály pro tkáňové náhrady. Bioaktivní povrchy
takovýchto materiálů mohou být následně připraveny povrchovou
dekompozicí amfifilních blokových kopolymerů1. Důležitých
biomimetických vlastností může být dosaženo užitím kopolymerů
obsahujících peptidové sekvence odvozné z fibronektinu a lamininu (jako
například GRGDSG, PHSRN, REDVDY, SIKVAVS a YIGSR). Alifatické
polyestery získané polymerací laktonů jako polylaktid (PLA),
polyglykolid (PGA), e-polycaprolakton (PCL) jsou často využívány pro
přípravu trojrozměrných scaffoldů v tkáňovém inženýrství.
V předkládaném příspěvku je diskutován způsob syntézy amfifilních
blokových kopolymerů tvořených hydrofobními polylaktidovými a
hydrofilními polyethylenoxidovými (PEO) bloky nesoucími proměnlivé
oligopeptidové sekvence umístěné na konci PEO bloku a umožňující řízení
specifické a nespecifické buňečné adheze.
Plně chráněné peptidové fragmenty byly připraveny syntézou na pevné
fázi Fmoc strategií s užitím chlorotritylové pryskyřice a následným
odstraněním z nosiče za použití 25% HFIP roztoku v CH2Cl2.
Kopolymer peptid-polyethylenoxid byl připraven couplingem aktivovaného
chráněného peptidového fragmentu s amino-PEO v prostředí DMF.
Polylaktidový blok byl následně naroubován řízenou polymerací laktidu
iniciovanou koncovou hydroxylovou skupinou PEO konjugátu. Cílový peptid-block-PEO-block-PLA kopolymer byl následně získán odstraněním chránících skupin peptidu.
References
1. Kubies, D., Rypáček, F., Kovářová, J., and Lednický, F., Microdomain structure in polylactide-block-poly(ethylene oxide) copolymer films, Biomaterials 21, 529, 2000.
2. Proks V., Machová L., Popelka Š and Rypáček F.
Biodegradable copolymers carrying cell-adhesion peptide sequences in Adv. Exp. Med. Biol, , 534: 191. 2003.
MODIFIKACE TROJROZMĚRNÝCH POLYMERNÍCH STRUKTUR BIOAKTIVNÍMI MOTIVY
R. HOBZOVÁ, H. KOMPERTOVÁ, J. MICHÁLEK, V. GURYČA, M. PŘÁDNÝ, B. DVOŘÁNKOVÁ
Ústav makromolekulární chemie Akademie věd České republiky, Heyrovského nám. 2, Praha 6
V posledních desetiletích došlo k velkému rozvoji tkáňového
inženýrství, včetně buněčných technologií, kterých lze využít mimo jiné
pro regeneraci a náhradu poškozených tkání. Vhodnými kandidáty pro
cílenou kultivaci buněk se zdají být syntetické hydrogely, které se
některými svými vlastnostmi blíží vlastnostem tkáně (např. obsahem
vody, mechanickými vlastnosti nebo biokompatibilitou). Zároveň lze řadu
jejich fyzikálně-chemických vlastností kontrolovat.
Je známo (Smetana 1993), že pro adhezi a proliferaci buněk na
povrchu biomateriálu je žádoucí přítomnost biologicky aktivních molekul
(proteinů, glykoproteinů, sacharidů). K zavedení bioaktivních motivů
byl zvolen komplex avidin-biotin, který reprezentuje jednu z
nejsilnějších nekovalentních vazeb. Vhodný bioaktivní motiv je začleněn
do polymerní struktury reakcí jeho biotinylované formy s avidinem
imobilizovaným na povrchu polymerního nosiče a vznikem pevného komplexu
avidin-biotin. Avidin je možné imobilizovat na povrch nosiče pomocí
nespecifických interakcí, elektrostatických sil nebo kovalentních
vazeb. Kovalentní vazba avidinu vyžaduje přítomnost reaktivních skupin
na povrchu polymeru, což je možné provést podle reakcí (Hermanson 1996,
Lomant a kol. 1976, aj.). Pro přípravu polymerních nosičů byly použity
kopolymery na bázi 2‑hydroxyethylmethakrylátu a kyseliny methakrylové s
různým stupněm hydrofility. Ke kultivaci byly použity linie lidských
keratinocytů a myších fibroblastů.
V této práci byly porovnány různé způsoby imobilizace avidinu a
prokázána účinnost vybraných bioaktivních motivů pro kultivace
některých typů buněk v sériích polymer, polymer + avidin, polymer +
avidin + biotinylovaný set.
Autoři děkují Grantové agentuře Akademie věd ČR za finanční podporu poskytnutou v rámci projektu č. 1QS400500558.
MULTIBLOKOVÉ POLYMERY POLY(ETHYLENGLYKOLU) PRO BIOMEDICINÁLNÍ APLIKACE
M. PECHAR1, A. BRAUNOVÁ1, K. ULBRICH1, M. JELÍNKOVÁ2, B. ŘÍHOVÁ2, L. SEYMOUR3
1Ústav makromolekulární chemie, AV ČR, 162 06 Praha 6, Česká republika; e-mail: pechar/imc.cas.cz
2Mikrobiologický ústav, AV ČR, 142 20 Praha 4, Česká republika
3Dept. of Clinical Pharmacology, Oxford University, Radcliffe Infirmary, Oxford, UK
Souhrn
Byla připravena a charakterizována řada nových
biodegradovatelných polymerů na bázi poly(ethylenglykolu) (PEG),
obsahujících enzymově, redukčně a hydrolyticky štěpitelné vazby. Tyto
polymery byly navrženy a studovány za účelem modifikace
nízkomolekulárních léčiv, proteinů a vektorů pro genovou terapii.
Ukázali jsme, že připojením k těmto polymerům mohou biologicky aktivní
látky výrazně zlepšit své farmakokinetické vlastnosti a snížit
nežádoucí toxicitu. Biodegradovatelnost polymerních nosičů zajistí
jejich snadné vyloučení z organismu ve formě degradačních produktů.
Úvod
Léčiva, proteiny nebo vektory pro dopravu genů modifikované biokompatibilními polymery jako jsou např. deriváty N-(2-hydroxypropyl)methakrylamidu
[1] či PEG [2] mohou výrazně zlepšit své farmakokinetické vlastnosti a
snížit nežádoucí toxicitu. Za předpokladu, že modifikující
makromolekuly jsou biodegradovatelné, je možné použít pro terapeutické
aplikace dokonce i vysokomolekulární polymery, neboť jejich degradační
produkty se nakonec vyloučí glomerulární filtrací. Důvody pro použití
vysokomolekulárních látek jsou, zejména u protinádorových léčiv, jejich
zvýšená akumulace v nádorové tkáni v důsledku tzv. EPR efektu (enhanced
permeability and retention effect) [3]. V této práci jsou prezentovány
multiblokové polymery na bázi PEG obsahující enzymově, redukčně a
hydrolyticky štěpitelné vazby.
Experimentální metody
Enzymově a hydrolyticky štěpitelné multiblokové
polymery byly připraveny polykondenzací diaminů na bázi oligopeptidů s
PEG bis(sukcinimidyl karbonátem) (PEG-BSC). Oligopeptidové bloky N,N'-bis(glutamyl)lysin (EKE) [4] a N,N'-bis(aspartylprolyl)lysin ((DP)2K)
[5] byly získány pomocí manuální peptidové syntézy na pevné fázi.
Protinádorové léčivo doxorubicin (Dox) bylo kovalentně navázáno na
polymer buď prostřednictvím enzymově štěpitelné tetrapeptidové spojky
GFLG [6], nebo acidolabilní hydrazonovou vazbou [7]. Polymery
aktivované zavedením 4-nitrofenylových skupin (Schéma 1) byly použity
pro povrchovou modifikaci virálních i nevirálních vektorů pro transport
genů. Degradace polymerního nosiče a uvolňování léčiva jak enzymovou,
tak chemickou hydrolýzou byly sledovány pomocí rozměrově vylučovací
chromatografie (SEC).
Polymery s redukčně štěpitelnými disulfidovými
vazbami v hlavním polymerním řetězci byly připraveny reakcí PEG-BSC s S-trityl-L-cysteinem
následovanou oxidační polykondenzací vzniklého derivátu PEG-cysteinu
jódem. Nakonec byly karboxylové skupiny vzniklého polymeru převedeny na
reaktivní sukcinimidylesterové skupiny (Schéma 2). Aktivovaný polymer
byl využit pro modifikaci povrchu polyelektrolytového komplexu
poly(lysin)/DNA sloužícího jako vektor pro dopravu genetické informace
do buňky. Schopnost polymerů podléhat redukční degradaci glutathionem
(GSH) byla studována pro různé koncentrace GSH (poměr GSH:S-S byl 5:1,
2:1 a 1:1, nejvyšší koncentrace byla 3×10-3 M). Hydrolytická
stabilita PEG-cystinu byla ověřena ve fosfátových pufrech o pH 8,0; 7,4
and 5,5. Uvolňování výchozího PEG 2000 bylo sledováno pomocí SEC.
Výsledky a diskuse
Polykondenzace PEG-BSC s oligopeptidy-diaminy EKE a (DP)2K vedla ke vzniku multiblokových polymerů o molekulové hmotnosti Mw = 40 000 a polydisperzitě Mw/Mn=2,5. Obě peptidové sekvence použité jako spojky mezi bloky PEG podléhaly enzymové hydrolýze katepsinem B, pentapeptid (DP)2K
byl dokonce štěpitelný chemickou hydrolýzou při pH 5,5. Tato skutečnost
může zajistit případnou degradaci polymerů v živých buňkách a následné
vyloučení metabolitů z organismu. Polymerní konjugáty s Dox vykazovaly
vyšší protinádorovou aktivitu a nižší akutní toxicitu než volné léčivo.
V případě modifikace poly(lysin)/DNA komplexů a adenovirálních vektorů
připravenými multivalentními hydrofilními polymery byla zjištěna
prodloužená doba cirkulace, snížená sorpce na krevní bílkoviny a
folátem směrovaná transgenní exprese [8].

Multiblokový polymer PEG-cystin (Mw=25
000) byl inkubován ve fosfátových pufrech pH 5,5; 7,4 a 8,0
odpovídajícím pH v endosomech, v krvi a v tenkém střevě. Za těchto
podmínek neprobíhala hydrolýza urethanových vazeb měřitelnou rychlostí.
Přítomnost přirozeného intracelulárního redukčního činidla GSH
(tripeptid Glu-Cys-Gly) vedla k velmi rychlému štěpení S-S vazeb.
Dokonce ani při nejvyšší použité koncentraci GSH (3×10-3 M)
nedošlo k úplnému rozštěpení všech vazeb. V systému se pravděpodobně
ustavila rovnováha mezi oxidovanými a redukovanými formami GSH a
polymeru.
Závěr
Navázání Dox na polymerní nosiče s oligopeptidovými spojkami EKE a (DP)2K
vedlo ke zvýšení terapeutické aktivity a snížení toxicity konjugátů ve
srovnání s volným léčivem. Modifikace jak virálních, tak nevirálních
vektorů pro dopravu genů reaktivními multivalentními polymery umožnilo
prodlouženou cirkulaci vektorů a jejich směrování ke specifickým
receptorům. Nové vysokomolekulární polymery na bázi PEG-cystinu,
obsahující urethanové a disulfidové vazby, jsou snadno štěpitelné
pomocí GSH. Tyto polymery jsou v současné době testovány jako nosiče
proteinů, vektorů pro genovou terapii a nízkomolekulárních léčiv.
Literatura
[1] J. Kopeček, P. Kopečková, T. Minko, Z. R. Lu, Eur. J. Pharm. Biopharm. 50 (2000) 61-81.
[2] S. Zalipsky, Adv. Drug Delivery Rev. 16 (1995) 157-182.
[3] H. Maeda, Y. Matsumura, Crit. Rev. Ther. Drug Carrier Syst. 6 (1989) 193-210.
[4] M. Pechar, K. Ulbrich, V. Šubr, L. W. Seymour, E. H. Schacht, Bioconjugate Chem. 11 (2000) 131-139.
[5] M. Pechar, A. Braunová, K. Ulbrich, Collect. Czech. Chem. Commun. 70 (2005) 327-338.
[6] M. Pechar, K. Ulbrich, M. Jelínková, B. Říhová, Macromol. Biosci. 3 (2003) 364-372.
[7] M. Pechar, A. Braunová, K. Ulbrich, M. Jelínková, B. Říhová, J. Bioact. Compat. Polym. 20 (2005) 319-341.
[8] C. M. Ward, M. Pechar, D. Oupický, K. Ulbrich, J. Gene Medicine 4 (2002) 536-547.
Poděkování
Práce byla finančně podporována grantem MŠMT ČR č. IM4635608802,
grantem Grantové agentury Akademie věd České republiky č. IAA4050201 a
grantem EU GIANT č. 512087.
POLYMERNÍ KONJUGÁT DEXAMETHASONU S ŘÍZENÝM UVOLŇOVÁNÍM LÉČIVA
H. KRAKOVIČOVÁ, T. ETRYCH, K. ULBRICH
Ústav makromolekulární chemie, Akademie věd České republiky, Heyrovského nám. 2,
162 06 Praha 6, Česká republika (krakovicova/imc.cas.cz)
ÚVOD
|

|
| Obr. 1: Schéma struktury konjugátu dexamethasonu |
Dexamethason
(DEX) patří do skupiny syntetických glukokortikoidů, odvozených od
přirozených hormonů vytvářených v kůře nadledvin. DEX se používá v
běžné praxi při léčbě mnohočetného myelomu nebo hormondependentních
nádorů (nádory prsu, prostaty) v kombinaci s jinými cytostatiky např.
paclitaxelem nebo doxorubicinem1. Má také antiedematosní,
protizánětlivé a imunosupresivní účinky. Uvedené účinky léčiva
dexamethason ukazují na jeho široké použití v humánní medicíně.
V naší laboratoři se zabýváme přípravou vodorozpustných kopolymerů na bázi N-(2-hydroxypropyl)methakrylamidu
(HPMA), vhodných jako nosiče biologicky aktivních látek. V nedávné době
jsme popsali nové typy kopolymerů HPMA s
6-(methakryloylamino)-hexanoylhydrazidem (MA-AH-NHNH2)2,
které představují vhodné polymerní nosiče pro cílenou dopravu léčiva
doxorubicinu navázaného na zmíněný polymerní nosič přes
hydrolyzovatelnou hydrazonovou vazbu3. Na základě
experimentálních výsledků předpokládáme, že hydrazonová vazba bude
stabilní v krevním řečišti (pH=7,4) a léčivo bude uvolněno v aktivní
formě díky poklesu pH teprve po vstupu do endozomů a lysozomů
nádorových buněk (pH=5 ‑ 6)4.
V této práci prezentujeme nový typ polymerních konjugátů s léčivem
dexamethason (Obr.1). Derivatizovaný dexamethason je na polymerní nosič
na bázi kopolymeru HPMA navázán pomocí hydrolyzovatelné hydrazonové
vazby. Vazbou DEX na polymerní nosič by mělo být dosaženo zvýšení
rozpustnosti hydrofobního léčiva, prodloužení doby cirkulace v krvi,
řízeného uvolňování léčiva v organismu a zajištění lepší biologické
dostupnosti.
VÝSLEDKY A DISKUZE
Příprava polymerního konjugátu dexamethasonu sestává ze tří
syntetických kroků. V prvním kroku je radikálovou kopolymerizací
monomerů HPMA a MA-AH-NHNH2 s iniciátorem
(2,2'-azobis-isobutyronitril) připraven reaktivní kopolymer - polymerní
nosič obsahující hydrazidové skupiny. Molekulová hmotnost nosiče může
být řízena koncentrací monomerů, iniciátoru, a nebo polymerizační
teplotou. V druhém kroku je dexamethason derivatizován na primární
hydroxylové skupině kyselinou levulovou (4-oxopentanovou) a karbonylová
skupina kyseliny levulové je ve třetím kroku využita k vazbě
derivatizovaného dexamethasonu na připravený polymerní nosič
pH-senzitivní hydrazonovou vazbou. Výsledky uvolňování léčiva z
kopolymeru o Mw ~ 34 000 a obsahu 8,4 váh. % DEX sledovaného
v in vitro podmínkách při 37 °C prokázaly, že polymerní
konjugát dexamethasonu je při inkubaci ve vodném pufru o pH 7,4
modelujícím pH krevního řečiště poměrně stabilní a dochází k uvolnění
jen cca 20 % léčiva/24 h. Ve vodném pufru o pH 5, který simuluje pH
endosomu, případně lysozomu buňky, dojde v první fázi k hydrolytickému
uvolnění esteru kyseliny levulové z polymerního konjugátu. Dexamethason
pak vzniká následnou hydrolýzou esterové vazby mezi DEX a kyselinou
levulovou. Tímto způsobem dochází k uvolnění skoro 100 % DEX/24 h, což
je výrazně více, nežli se uvolní v prostředí modelujícím podmínky při
transportu polymerního léčiva organismem.
Zkušeností s přípravou konjugátu HPMA kopolymeru a dexamethasonu
bude v budoucnu využito pro přípravu polymerních léčiv s duálním
účinkem, tj. polymerních konjugátů nesoucích na témže polymerním
řetězci dva typy léčiv s doplňujícím se, případně i synergickým účinkem.
Poděkování: Tato práce byla podporována GA AV ČR (grant č. A4050201) a grantem MŠMT ČR
(č. IM 4635608802).
Literatura:
- Skommer J; Wlodkowic, D; Matto, M; Eray, M; Pelkonen, J. Leukemia Research 2006, 30, 322-331.
- Etrych, T; Jelínková, M; Říhová, B; Ulbrich, K. J. Controlled Release 2001, 73, 89.
- Etrych, T; Chytil, P; Jelínková, M; Říhová, B; Ulbrich, K. Macromol. Biosci. 2002, 2, 43
- Ulbrich, K; Etrych, T; Chytil, P; Jelínková, M; Říhová, B. J. Controlled Release 2003, 87, 33.
POVRCHOVÁ MODIFIKACE POLYELEKTROLYTOVÝCH KOMPLEXŮ DNA/PLL
L. KOSTKA, V. ŠUBR, R. LAGA, K. ULBRICH
Department of Biomedicinal Polymers, Institute of Macromolecular
Chemistry, Academy of Sciences of the Czech Republic, Heyrovského nám.
2, CZ-162 06 Praha 6, Czech Republic (kostka/imc.cas.cz)
Genová terapie se stává jednou z nejintenzivněji se rozvíjejících
strategií výzkumu vývoje nových léčiv. V humánní medicíně nabízí nové
léčebné možnosti pro mnoho běžných i dědičných nemocí, pro které jsou
konvenční klinické metody málo účinné. Mezi takové nemoci lze řadit
monogenické poruchy, cystickou fibrózu, ale také mnohem více komplexní
nemoci, jako kardiovaskulární nemoci, poruchy nervového nebo
autoimunitního systému či nádorová onemocnění.
V případech nemocí způsobených mutací specifických genů nebo jejich
produktů nabízí genová terapie doručení funkční kopie tohoto genu do
cílové buňky či orgánu a tím dosažení terapeutického účinku. Nicméně
může to být také nástroj pro léčení negenetických či polygenických
poruch doručením genů stimulujících odpověď imunitního systému, genů
modifikujících buněčné informace, podporujících buněčný vývoj, či genů
produkujících terapeutické proteiny specifických funkcí.
Bohužel, volná DNA není vhodná pro dopravu genetického materiálu in vivo
kvůli degradaci sérovými nukleasami a malé specifitě účinku. Je proto
nutné použít vhodný dopravní systém. Ideální dopravní systém by měl být
stabilní v krevním řečišti, ochránit DNA před degradací během
transportu, zabránit zachycení složkami retikuloendoteliálního systému
(RES), být dostatečně malým pro extravazaci a být buněčně či orgánově
specifický. Po dosažení cílového orgánu by měl dopravní systém
proniknout do buňky (endocytózou), zabránit interakci DNA s
lysozomálnímy enzymy a dopravit ji do buněčného jádra. Je zřejmé, že
ideální dopravní systém musí obsahovat více složek zodpovědných za
specifické chování celého vektoru.
V principu jsou vyvíjeny dva základní nosičové systémy, virální
(adeno- a retrovirální) a nevirální (syntetické). S použitím virálních
vektorů je v klinické praxi spojeno několik vážných problémů - riziko
imunitní odpovědi organismu na virální částice, riziko náhodné
integrace zprostředkované viry nebo jejich rekombinace s jiným typem
viru. Tato práce se zabývá druhým typem dopravního systému, tzv.
nevirálních vektorů na bázi polyelektrolytových komplexů.
Byla studována příprava a povrchová modifikace polyelektrolytových
komplexů DNA s polykationty (PEK), navržených jako nevirální vektory
pro dopravu genové informace in vivo. PEK byly připraveny
samouspořádáním hydrofilního polykationtu a DNA ve vodném roztoku.
Všechny PEK byly připraveny s poměrem nábojů (+:- = 2:1). Takto
připravený komplex je sice schopen ochránit DNA (nebo DNA plasmid
kódující určitý gen) před degradací v průběhu transportu, interaguje
však s krevními proteiny a je zachycován mikrofágy i buňkami RES. Proto
je nezbytné takový komplex určený pro in vivo aplikaci
povrchově upravit a současně zavést na jeho povrch skupiny schopné
zajistit specifickou dopravu komplexu k vybraným buňkám. Povrchová
modifikace (coating) byla uskutečněna pomocí hydrofilních kopolymerů na
bázi N-(2-hydroxypropyl)metakrylamidu (HPMA) o kterých je
známo, že snižují imunogenicitu a zlepšují biokompatibilitu tímto
polymerem modifikovaných biologicky aktivních molekul. Pro účely
povrchové modifikace DNA vektorů jsme vyvinuli nové reaktivní
kopolymery obsahující reduktivně štěpitelnou disulfidickou vazbu. Tato
vazba by měla umožnit uvolnění polymeru z povrchu částice v reduktivním
prostředí cytoplasmy po jejím průniku do buňky a tím usnadnit uvolnění
DNA z komplexu v cílové buňce.
Pomocí rozptylu světla byla sledována molekulová hmotnost (Mw) a hydrodynamický poloměr (RH) komplexu, stabilita PEK byla sledována pomocí UV spektroskopie a horizontální elektroforézy.
Příprava modifikovaných PEK zahrnuje tři syntetické kroky. V prvním
kroku je roztokovou radikálovou kopolymerací monomerů HPMA a Ma-AH-TT,
resp. HPMA a Ma-GlyGly-TT (TT je reaktivní acylthiazolidin-2-thionová
skupina) s iniciátorem 2,2´-azobisisobutyronitrylem připraven kopolymer
obsahující reaktivní TT skupiny schopné za mírných podmínek reagovat s
primární aminoskupinou, které jsou v druhém kroku polymeranalogickou
reakcí s S-(2-pyridylthio)cysteamine hydrochloridem převedeny na
pyridyldisulfidové skupiny. Třetím krokem syntézy je modifikace části
aminoskupin poly(L-lysinu) 2-iminothiolanem, tzn. nahrazení těchto
ε-aminoskupin thiolovými skupinami. Ve finální části syntézy reaguje
reaktivní HPMA kopolymer s -SH skupinami modifikovaným DNA komplexem za
tvorby disulfidových můstků mezi komplexem a hydrofilním polymerem.
V průběhu modifikace došlo k nárůstu Mw a RH, velikost
částic však i po modifikaci vyhovuje požadavkům na úspěšnou
extravasaci. Modifikované PEK byly přečištěny pomocí HPLC. Modelová
inkubace modifikovaných PEK s L-glutathionem vedla k rozštěpení
disulfidické vazby a uvolnění polymeru z povrchu komplexu, což se
projevilo poklesem Mw a RH. Došlo také k uvolnění DNA, což bylo potvrzeno pomocí horizontální elektroforézy.
Poděkování: Tato práce byla podporována grantem EU, GIANT, č. 512087 a firmou Zentiva a.s.
VPLYV TVORBY FYZIKÁLNEJ SIETE NA REOLOGICKÉ VLASTNOSTI BIODEGRADOVATEĽNÝCH POLYMÉRNYCH
D. MOŠKOVÁ, I. CHODÁK
Ústav polymérov, Slovenská Akadémia Vied (Centrum excelentnosti
CEDEBIPO), Dúbravská cesta 9, 842 36 Bratislava, Slovensko, e-mail:
upoldamo/savba.sk
Biodegradovateľné polyméry zaznamenávajú v súčasnosti veľkú
pozornosť ako materiály vhodné na zredukovanie enviromentálnych
problémov s plastovým odpadom. Pri ich aplikácii je však dôležité
zamerať sa na zlepšenie niektorých nevyhovujúcich úžitkových
vlastností, ktoré je do určitej miery možné odstrániť aplikáciou
stužujúcich plnív. Klasifikácia biodegradovateľných materiálov v zmysle
ISO 14855 pripúšťa maximálny obsah nebiodegradovateľných prísad do 5
hm. %. Z tohto dôvodu sme zamerali pozornosť na nanokompozity s
vrstevnatými silikátmi, pri ktorých už pomerne malé množstvo plniva v
polymérnej matrici spôsobí výrazné zlepšenie termických a mechanických
vlastností.
Nanokompozity na báze biodegradovateľnej polymérnej matrice
(polykaprolaktón - PCL) a vrstevnatých silikátov (organomodifikované a
prírodné Cloisity) sa pripravili miešaním v tavenine. Na základe
sledovania mechanických vlastností sa stanovil optimálny obsah
organomodifikovaného Cloisitu 20A 3 hm.%, zatiaľ čo prídavok
nemodifikovaného Cloisite Na sa na mechanických vlastnostiach
neprejavil pozitívne.
Reologické vlastnosti sa analyzovali na základe viskoelastických
vlastností z oscilačného reologického merania. Viskoelastické
vlastnosti kompozitov s organomodifikovaným plnivom sa posunuli do
oblasti "solid-like" v dôsledku prekročenia reologického perkolačného
prahu a následnej tvorby fyzikálnej siete. V nízkofrekvenčnej oblasti
dochádza k výraznému nárastu modulov a ich relatívne nízka závislosť od
frekvencie poukazuje na silné interakcie medzi reťazcami polyméru a
jednotlivými časticami plniva. S rastúcim obsahom plniva klesá hodnota
smernice G´ a G´´ a crossover point (G´= G´´)
sa posúva do oblasti nižších frekvencií v dôsledku koexistencie
interakcií častica - častica a častica - polymér a rovnako v dôsledku
dosiahnutia kritickej koncentrácie plniva, nad ktorou dochádza ku
vzniku sekundárnej sieťovej štruktúry. Nanokompozity vykazujú vyššiu
komplexnú viskozitu v porovnaní s matricou a výrazné "shear thinning" v
oblasti nízkych frekvencií. Štruktúrne zmeny v závislosti od
koncentrácie plniva sa stanovili pomocou Cole-Cole diagramov. Z grafov
vidno odchýlku kriviek kompozitov od krivky polymérnej matrice v
dôsledku štruktúrnych zmien v kompozite. S nárastom obsahu plniva sa
stupeň odchýlky zvyšuje. Pravdepodobne dochádza k tomuto javu v
dôsledku výrazného nárastu interakcií častica - častica s nárastom
obsahu plniva, spôsobených klesajúcou vzájomnou vzdialenosťou medzi
jednotlivými časticami plniva.
Táto práca bola podporovaná Agentúrou na podporu výskumu a vývoja na základe
Zmluvy č. APVV-51-050505
BIOLOGICKY ROZLOŽITELNÉ POLYMERY
D. CHROMCOVÁa, J. NÁHLÍKb, V. ŠAŠEKc, J. RODAa, J. BROŽEKa
aÚstav polymerů, bÚstav inženýrství pevných
látek, Vysoká škola chemicko-technologická v Praze, Technická 5, 166 28
Praha 6, Česká republika (Jiri.Brozek/vscht.cz).
cMikrobiologický ústav AV ČR, Vídeňská 1083, 142 20 Praha 4, Česká republika
Syntetické polymery jsou v dnešní době nepostradatelnými materiály v
různých odvětvích lidské činnosti. Důsledkem nárůstu spotřeby
polymerních materiálů je na druhé straně vznik velkého množství odpadu,
což má negativní dopad na životní prostředí. Proto se pozornost
výzkumných pracovníků soustřeďuje na vývoj nových biologicky
rozložitelných polymerních materiálů. Tyto materiály by v některých
aplikacích mohly nahradit část prakticky nebiodegradovatelných
materiálů.
Naše výzkumná aktivita je zaměřena na dva typy polymerních materiálů - (i) chemicky modi-fikovaný poly(3-hydroxybutyrát) a (ii) polyesteramidy na bázi e-kaprolaktamu a e‑kaprolaktonu.
Poly(R)-3-hydroxybutyrát) (PHB) je příkladem termoplastu, který je
produkován některými typy bakterií s obnovitelných zdrojů. Rozšíření
aplikačních možností PHB brání problém s jeho fyzikálním stárnutím
(křehnutím) a i jeho nízká tepelná stabilita v blízkosti jeho teploty
tání. Zavedením vhodných strukturních jednotek do PHB makromolekul lze
výrazně ovlivnit chemickou strukturu, porušit stereoregularitu PHB
řetězců a získat tak materiál s příznivějšími vlastnostmi než
nemodifikovaný PHB. K tomu lze využít transesterifikačních reakcí mezi
PHB a jinými polyestery. Vhodným polyesterem by mohl být
poly(e-kaprolakton), který též náleží do skupiny biologicky
rozložitelných materiálů. V literatuře popsané transesterifikační
reakce byly prováděny v rozpouštědlech, která jsou z hygienického
hlediska nevhodná. Náš přístup spočíval v modifikaci PHB
poly(e‑kaprolaktonem), přičemž v první fázi procesu e-kaprolakton (CLO)
sloužil jako rozpouštědlo pro PHB. Sloučeniny, které byly použity k
iniciaci polymerace CLO byly voleny tak, aby katalyzovaly i výměnné
(transesterifikační) reakce. Tímto způsobem získané materiály
vykazovaly podle DSC měření dva endotermy tání odpovídající
poly(e-kaprolakonu) (PCLO) a modifikovanému PHB. Pokles teploty tání
modifikovaného PHB svědčí o podílu transesterifikačních reakcí mezi in situ vytvářeným PCLO a PHB.
Anionovou kopolymerací e-kaprolaktamu (CLA) a e-kaprolaktonu (CLO)
byly připraveny polyesteramidy (PEA). Technikou polymeračního odlévání
byly připraveny PEA s obsahem laktonových strukturních jednotek v
rozsahu 10 až 100 mol%. S rostoucím obsahem CLO strukturních jednotek
(v rozmezí 10 -60 mol%) zabudovaných v PEA klesá teplota tání
kopolymeru, obsah krystalické fáze, modul pružnosti v tahu a pevnost
při přetržení vzorku. Testy abiotické hydrolýzy prováděnými ve
fosfátovém pufru při pH=7,4 a teplotě 60 °C bylo zjištěno, že s obsahem
strukturních jednotek CLO v PEA roste citlivost kopolymeru k hydrolýze
indikované úbytkem hmotnosti a snížením redukované viskozity (ηred) u degradovaných vzorků.
Biologická rozložitelnost PEA byla ověřována jednak pomocí
kompostovacího testu, jednak sledováním vlivu extracelulární
enzymatické aktivity vybraných kmenů ligninolytických hub. Po 100 dnech
kompostování výrazně klesá ηred roztoků PEA vzorků, v
případě kopolymeru s obsahem laktonových strukturních jednotek 60mol%
CLO dokonce na hodnoty méně než poloviční ve srovnání se vzorkem
výchozím. Probíhající degradace v polymeru dokresluje výrazné narušení
povrchů testovaných fólií. Tentýž polymer byl po 63 dnech kompostování
za řízených podmínek, při konstantní teplotě 60 °C desintegrován. Změny
redukovaných viskozit ηred roztoků PEA vystavených působení
hub byly ve srovnání se změnami roztokových vlastností polymerů po
abiotické hydrolýze jen malé. Relativně nejvýraznější pokles ηred
byl zaznamenán u polyesteramidů s nejvyšším obsahem laktamové složky.
Obdobné testy lze uplatnit při studiu biodegradace modifikovaného PHB
Tato práce vznikla za podpory grantu č. 203/06/0528 GA ČR a výzkumného záměru MSM 6046137302.
SMĚSI AROMATICKO-ALIFATICKÉHO KOPOLYESTERU SE ŠKROBEM
I. PROKOPOVÁ, P. NIKLOVÁ, J. ŠIMEK
Ústav polymerů, Vysoká škola chemicko-technologická v Praze,
Technická 5, 166 28 Praha 6, Česká republika, e-mail:
irena.prokopova/vscht.cz
Škrob, jeden z nejlevnějších biodegradovatelných polymerů, izolovaný
přímo z obnovitelných zdrojů, vzbudil pozornost jako potenciální
surovina pro technické aplikace, zvláště pro výrobu obalů, již před
více než třemi desítkami let. Samotný plastifikovaný škrob, označovaný
také jako termoplastický škrob, má však v důsledku vysoké hydrofility a
malé mechanické pevnosti limitované využití. Jednou z možností
překonání těchto nedostatků, při současném zachování biologické
rozložitelnosti materiálu, je příprava blendů škrobu se syntetickými
biodegradovatelnými polymery. Vedle polyvinylalkoholu, jehož směsi se
škrobem byly patentovány již v sedmdesátých letech minulého století
[1], k tomu lze využít alifatické polyestery, např. poly(e-kaprolakton)
nebo kyselinu polymléčnou, polyesteramidy, případně kopolyestery
obsahující aromatické strukturní jednotky. Vhodné pro přípravu
podobných blendů jsou i bakteriální (ko)polyestery -
poly(hydroxyalkanoáty) [2].
V naší práci jsme se zaměřili na přípravu a charakterizaci směsí
termoplastického škrobu s alifaticko-aromatickým kopolyesterem na bázi
polyethylentereftalátu (PETP) a kyseliny mléčné. Kopolyester byl
připraven polykondenzací produktu solvolýzy PETP z použitých nápojových
lahví vodným roztokem kyseliny mléčné provedené při 250°C za katalýzy
octanem zinečnatým. Biologická rozložitelnost tohoto typu kopolyesteru
byla již dříve prokázána pomocí kompostovacího testu [3]. Pro přípravu
směsí se škrobem byl použit kopolyester obsahující podle analýzy NMR 35
mol% aromatických strukturních jednotek (hred jeho roztoku v trikresolu je 60 cm3/g). Podle termické analýzy má kopolyester amorfní strukturu s teplotou skelného přechodu 67 °C.
Nativní pšeničný škrob s obsahem 12 hmot.% vody byl bezprostředně
před smísením s kopolyesterem destrukturizován při 130°C v přítomnosti
15 - 30 hmot.% glycerolu v hnětiči Brabender (PLASTI-CORDER
LAB-STATION) při 50 otáčkách/min. Za stejné teploty a podmínek hnětení
byl připravený termoplastický škrob (TPS) smísen s kopolyesterem v
hmotnostních poměrech 25:75, 35:65, 45:55. Ze směsí byly lisovány fólie
pro přípravu zkušebních tělísek. Obsah glycerolu ve fóliích, stanovený
HPLC analýzou jejich vodných extraktů, odpovídá v rámci chyby stanovení
obsahu glycerolu dávkovaného k přípravě TPS. Při 66% relativní vlhkosti
a 20°C byla stanovena sorpce vody směsí. Její hodnota se pohybuje, jak
je zřejmé z Obr. 1, zhruba v rozmezí 4-8 %, v závislosti na složení
směsi. Smáčivost povrchu fólií, indikovaná kontaktním úhlem, který s
povrchem fólie svírá přisedlá kapka vody, logicky vzrůstá s rostoucím
obsahem TPS ve směsi. Zjištěné změny charakteristických geometrických
parametrů přisedlé kapky vody - kontaktní úhel a "délka" kapky - byly
ve sledovaném časovém rozmezí 0 - 10 minut až překvapivě malé.
Přestože snímky kryogenních lomových ploch vzorků směsí
kopolyesteru s TPS ukázaly nepříliš dobrou adhezi mezi fázemi
syntetického a přírodního polymeru, byla u směsí (kondicionovaných při
66% rel. vlhkosti) prokázána dobrá pevnost v tahu (Obr. 2). Tažnost
směsí je však velmi nízká, nepřesahuje hodnoty jednotek procent.
Nejvyšší pevnost v tahu vykazují vzorky směsí s poměrem glycerol/škrob
0,25, tj. směsí připravených z TPS s obsahem 20 hmot.% glycerolu.
Samotný použitý kopolyester je relativně křehký materiál. Jeho
mechanické vlastnosti nelze charakterizovat za podmínek srovnatelných s
charakterizací jeho směsí s TPS.

Obr.1 Sorpce vody směsí aromaticko-alifatického kopolyesteru a TPS při 66% relativní vlhkosti a 20°C.
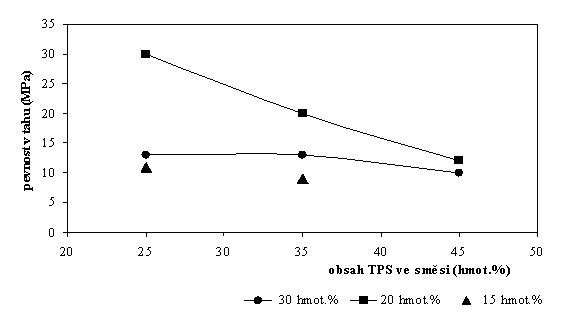
Obr. 2 Pevnost v tahu směsí aromaticko-alifatického kopolyesteru s
TPS. Zkušební tělíska kondiciována při rel. vlhkosti 66% a 20 °C.
Vlastnosti připravených směsí ukazují, že mísení
aromaticko-alifatického kopolyesteru na bázi PETP a kyseliny mléčné s
plastifikovaným pšeničným škrobem je možnou cestou přípravy biologicky
rozložitelného materiálu, který svými parametry může vyhovovat některým
aplikacím v obalové technice. Jeho mechanické vlastnosti lze nepochybně
ještě příznivě ovlivnit přísadou vhodného kompatibilizátoru.
Autoři děkují za podporu projektu Grantové agentuře České republiky
(Grant č. 203/06/0528) a MŠMT (Výzkumný záměr MSM 6046137302).
Literatura:
[1] Camerford J., Kapur Ch.: Franc. Pat. 2 308 656 (1976).
[2] Avérous L.: J. Macromol. Sci., Part C 44, 231 (2004).
[3] Prokopová I., Vlčková E., Skolil J., Šašek V., Musílková E.,
Náhlík J.: Kopolyestery na bázi polyethylentereftalátu a kyseliny
mléčné a jejich biologická rozložitelnost. Česko-slovenská konference
Polymery 2002. Praha 2002, P 18.
KOPOLYMERACE ε-KAPROLAKTAMU A ω-LAUROLAKTAMU
J. BUDÍN, J. RODA, J. BROŽEK
Ústav polymerů, Vysoká škola chemicko-technologická v Praze, Technická 5, 166 28 Praha 6,
Česká republika (budinj/vscht.cz, http://www.vscht.cz)
Polyamidy jsou známy od poloviny minulého století jako kvalitní
vláknotvorné materiály. Jedním z nich je polyamid 6, který je
připravován polymerací ε-kaprolaktamu. Tento plast se
vyznačuje řadou příznivých vlastností jako vynikající mechanickou
pevností, houževnatostí, odolností k abrazi a tepelnou stabilitou, díky
kterým se prosazuje také jako inženýrský plast. Tyto aplikace činí v
současné době okolo 30 % jeho spotřeby. Kromě řady zmíněných příznivých
vlastností má polyamid 6 také negativní vlastnosti a to především nižší
vrubovou houževnatost při záporných teplotách a vyšší rovnovážnou
sorpci vody. Pro další aplikační možnosti je nezbytné jeho vlastnosti
vhodně modifikovat. Existuje celá řada možností modifikace užitných
vlastností polyamidů: použití vhodných plniv, zabudování
polybutadienových nebo polyetherových řetězců do polymeru a v
neposlední řadě kopolymerace s laktamy o rozdílné velikosti cyklu.
Rozdílná délka methylenových sekvencí zapříčiňuje výrazné změny
vlastností díky snížení obsahu krystalické fáze. Kopolymerace laktamů
je atraktivní nejen z hlediska možnosti využit všechny reakční
mechanismy (hydrolytický, aniontový, kationtový) a jejich vlivu na
kinetiku a termodynamiku procesu, ale i možností modifikovat či
nastavit na míru vlastnosti polyamidů.
V předkládané práci jsme se zabývali porovnáním iniciačních systémů
při aktivované a neakti-vované kopolymerace ε-kaprolaktamu a ω-laurolaktamu. Jako iniciátory byly sledovány sodná sůl ε-kaprolaktamu a méně bazický ε-kaprolaktammagnesiumbromid, v případě aktivované kopolymerace byl kromě iniciátoru polymerace využit ještě aktivátor N‑benzoyl-ε-kaprolaktam.
Materiály připravené aktivovanou kopolymeraci (polymerační čas 30
minut, polymerační teplota 150°C) v závislosti na použitém iniciátoru
kopolymerace vykázaly fundamentální rozdíly ve vlastnostech. Materiál
připravený za iniciace sodnou solí ε‑kaprolaktamu měl
jeden endotherm tání ~130 - 140°C a dle NMR analýzy se jedná o
statistický kopolymer [1]. Naproti tomu materiál připravený za iniciace ε‑kaprolaktammagnesiumbromidem měl dva zřetelné
endothermy tání ~130 - 140°C a ~210°C a dle "frakcionace" vroucím
ethanolem (je známo, že statistický kopolymer ε‑kaprolaktamu a ω-laurolaktamu
je rozpustný ve vroucím ethanolu [2]) a NMR analýzy se jedná o směs
statistického kopolymeru a blokového kopolymeru obsahujícího bloky
statistického kopolymeru a poly(ε-kaprolaktamu) [1].
Byl sledován časový průběh kopolymerace ekvimolárního množství obou komonomerů. Při aktivované kopolymeraci v samém počátku polymerace vzniká v přítomnosti obou iniciátorů převážně poly(e-kaprolaktam) (endotherm tání ~215°C). Další průběh kopolymerace se však už významně liší. Při iniciaci sodnou solí ε-kaprolaktamu se v počátku kopolymerace vzniklý poly(ε-kaprolaktam)
rychle transformuje díky bazicky katalyzovaným výměnným reakcím v
statistický kopolymer s endothermem tání ~130 - 140°C. Při iniciaci s ε-kaprolaktammagnesiumbromidem
je průběh kopolymerace zcela odlišný. Vyšší endotherm tání zůstává
zachován během celého procesu polymerace. Nižší endotherm tání
odpovídající statistickému kopolymeru se objevuje po dosažení obsahu
polymeru ~50%. Toto rozdílné chování je pravděpodobně způsobeno
rozdílnou bazicitou obou iniciátorů a koordinací hořečnaté soli s
N-acyllaktamovými strukturami a tím snížením koncentrace soli
katalyzující výměnné reakce.
Zdánlivé rychlostní konstanty při neaktivované kopolymeraci ekvimolárního množství obou komononerů za iniciace sodnou solí ε-kaprolaktamu
leží mezi odpovídajícími homopolymeracemi. Bylo dosaženo ~95% obsahu
polymeru při všech polymeračních teplotách (220 -260°C) a teploty tání
kopírovaly obdobný trend jako při aktivované kopolymeraci. Zcela
odlišně se chovaly kopolymerace za iniciace ε‑kaprolaktammagnesiumbromidem.
Při všech polymeračních teplotách (220 - 280°C) byl dosažen obsah
polymeru jen ~30% a všechny endothermy tání odpovídaly teplotě tání
poly(ε‑kaprolaktamu).
Je patrné, že bazicita (disociace) ε-kaprolaktammagnesiumbromidu je výrazně nižší než sodné soli ε-kaprolaktamu, takže rovnováha mezi ε-kaprolaktamovými a ω‑laurolaktamovými anionty je posunuta ve prospěch ε-kaprolaktamového aniontu (aniontově aktivnějšího ε-kaprolaktamu), což způsobuje preferenční tvorbu poly(ε‑kaprolaktamu). V případě aktivované kopolymerace je výše zmíněná rovnováha pravděpodobně regulovaná přítomností aktivátoru.
Literatura
[1] Budín J., Roda J., Brožek J., Kříž J., Macromol. Symp. 2006, přijato.
[2] CZ 289909 (2002), M. Marek, I. Prokopová, J. Dvořáček, E. Vlčková, D. Čurda, CAN 141:125149.
Tato práce byla řešena jako součást výzkumného záměru MSM
6046137302 Příprava a výzkum funkčních materiálů a materiálových
technologií s využitím mikro- a nanoskopických metod.
Směsi poly(Vinylchloridu) a alifaticko-aromatických kopolyesterů na bázi recyklovaného poly(ethylentereftAlátu) a e-kaprolaktonu
R. Kalousková, D. Nevařil, I. Prokopová
Ústav polymerů, Vysoká škola chemicko-technologická v Praze,
Technická 5, 166 28 Praha 6, Česká republika (
Radka.Kalouskova/vscht.cz , http://www.vscht.cz/pol/)
Díky výborným mechanickým vlastnostem, snadné mísitelnosti s řadou
aditiv a zcela jistě i díky ceně se PVC stále řadí k nejrozšířenějším
polymerům. Aditiva , ať už nízko nebo vysokomolekulární jako např.
maziva, změkčovadla, plniva, modifikátory atd. usnadňují
zpracovatelnost směsí, rozšiřují aplikační možnosti materiálu a hrají
či nepochybně budou hrát roli i při jeho zhodnocení jako odpadu.
Míšení PVC s alifatickými, resp. aromatickými polyestery umožňují
specifické interakce mezi karbonylovou skupinou polyesteru a α-vodíkem
na řetězci PVC. Vedle interakčního parametru je však důležitým faktorem
ovlivňujícím mísitelnost polyesterů i poměr methylénových a
karbonylových skupin v polyesteru (CH2/COO)1,2. V
závislosti na tomto poměru prochází interakční parametr minimem a
vzniká tzv "okno mísitelnosti" ohraničené poměrem 3-14, přičemž bez
problémů jsou mísitelné polyestery s poměrem 4-10. Poly(e-kaprolakton)
má ideální poměr skupin (5) a vhodný interakční parametr ; patří proto
mezi polyestery nejlépe mísitelné s PVC . Na druhé straně
poly(ethylentereftalát) je vzhledem k nevyhovujícímu poměru CH2/COO skupin (2) a značnému příspěvku odpudivých sil ve směsi s PVC nemísitelný2.
Mísitelnost aromaticko-alifatických kopolyesterů na bázi
ethyléntereftalát/e-kaprolakton s PVC pak souvisí s podílem
e-kaprolaktonových strukturních jednotek v kopolyesteru3.
Příspěvek je úvodem k problematice směsí PVC a
alifaticko-aromatických kopolyesterů na bázi recyklovaného
poly(ethylentereftelátu) a e-kaprolaktonu. K míšení byly využity
kopolyestery syntetizované v ústavu polymerů z glykolyzátu odpadních
PET-lahví a e‑kaprolaktonu s nižší redukovanou viskozitou (0,15-0,44
100cm3/g)4. Podíl ethyléntereftalátových jednotek
v kopolyesteru činil 40- 60 mol%. Obsah kopolyesteru v blendu se
pohyboval v rozmezí 0-28 hm.%. Směsi připravené na dvouválci byly
hodnoceny z hlediska tepelné a světelné stability a mechanických
vlastností. Přídavek kopolyesteru ve směsi značně ovlivnil stabilitu
přítomného PVC; ta do určité míry závisí na typu použitého
stabilizačního systému. Např.v případě stabilizace podílu PVC
sloučeninami na bázi organocínu došlo ve srovnání se samotným PVC k
výraznému poklesu tepelné i světelné stability směsí, a to jak s
rostoucím obsahem kopolyesteru ve směsi, tak s rostoucím obsahem
etylentereftalátových jednotek v kopolyesteru. Změnou stabilizačního
systému byl tento trend potlačen.
Všechny směsi vykazovaly jednu hodnotu Tg, která s rostoucím obsahem
kopolyesterů klesala. Tvrdost směsí se ve srovnání se samotným PVC
téměř nezměnila, pevnost v tahu procházela maximem při obsahu cca 9hm.%
kopolyesteru ve směsi. Směsi především s vysokým obsahem kopolyesteru a
vyšším podílem ethylentereftalátových jednotek však postupem času
dokrystalizovávaly a křehly. Jak už bylo zmíněno, práce je zatím úvodem
k dané problematice a naznačuje směry dalšího studia, zejména v oblasti
použitých kopolyesterů, modifikace přípravy i složení výsledných směsí.
Literatura:
1.Woo E.M, Barlow J.W:. Polymer 26, 769 (1985),
2.Aubin M, Prud´homme R.E:. Polym. Eng. Sci 24, 350 (1984)
3.Ma De-Zhu, Prud´homme R.E:Polymer 31, 917 (1990)
4.Prokopová I., Vitásek J., Vlčková E, Náhlík J., Šašek V., APROCHEM 24.-27.4.2006 Milovy, Sborník přednášek 2, 2134
Tato práce vznikla za podpory výzkumného záměru MSM 6046137302.
HYDROLÝZA KOPOLYESTERŮ S AROMATICKÝMI STRUKTURNÍMI JEDNOTKAMI
J. VITÁSEKa, V. ŠAŠEKb, J. NÁHLÍKc, I. PROKOPOVÁa
a Ústav polymerů, cÚstav inženýrství pevných
látek, Vysoká škola chemicko-technologická v Praze, Technická 5, 166 28
Praha 6, Česká republika, e-mail: jiri.vitasek/vscht.cz
b Mikrobiologický ústav Akademie věd České republiky, Vídeňská 1083, 142 20 Praha 4,
Česká republika, e-mail: sasek/biomed.cas.cz
Aromatické polyestery, mezi nimiž má zvláště významné uplatnění
poly(ethylentereftalát) (PETP), se zařazují mezi polymery s mimořádnou
odolností k biologickému prostředí. Teprve před nedávnem se objevila
první práce [1], která ukazuje, že PETP relativně rychle hydrolyzuje
při 55°C účinkem hydrolázy z aktinomycety Thermobifida fusca.
Aromaticko-alifatické kopolyestery jsou naproti tomu členem
nepříliš početné skupiny syntetických, biologicky rozložitelných
polymerů. Lze je považovat za kompromis spojující velmi dobré
materiálové vlastnosti aromatických polyesterů s biologickou
rozložitelností alifatických polyesterů, kterým naopak pro aplikace
důležité mechanické a termické vlastnosti chybí. U řady typů
aromaticko-alifatických kopolyesterů jednak syntetizovaných
polykondenzací kyseliny tereftalové (nebo jejího dimethylesteru) a
různých alifatických dikarboxylových kyselin a diolů, a jednak
připravených transesterifikací aromatického a alifatického polyesteru,
byla prokázána degradace účinkem enzymů při 37°C, nebo účinkem
mikroorganismů v půdě či kompostu [2].
V této práci jsme se zaměřili na studium hydrolýzy
aromaticko-alifatických kopolyesterů připravených s využitím PETP z
použitých nápojových lahví. Byla sledována hydrolytická degradace
kopolyesterů v abiotickém prostředí při 60°C a pH 7 v závislosti na
poměru aromatických a alifatických -ε-kaprolaktonových - strukturních
jednotek v kopolyesterech. Citlivost kopolyesteru k mikrobiálně
katalyzované degradaci byla ověřována kompostovacím testem za
kontrolovaných podmínek (teplota, pH) a sledováním vlivu extracelulární
enzymatické aktivity vybraných kmenů ligninolytických hub. Změny ve
struktuře kopolyesterů vlivem abiotické hydrolýzy, nebo biologicky
aktivního prostředí, byly sledovány pomocí GPC a viskozimetrických
měření, a dále na základě porovnání termických vlastností původních a
degradovaných vzorků. U vybraných vzorků ve formě fólií či vláken byly
sledovány změny jejich povrchů pomocí SEM.
Bylo prokázáno, že v závislosti na obsahu aromatických strukturních
jednotek v kopolyesteru dochází v abiotickém prostředí při 60°C k
významnému statistickému štěpení makromolekul již během 4-6 týdnů. U
vzorků s obsahem aromatické složky nižším než 70 mol% byl zaznamenán
výrazný pokles molární hmotnosti po šestiměsíčním kompostování.
Autoři děkují za podporu projektu Grantové agentuře České republiky
(Grant č. 203/06/0528) a MŠMT ČR (Výzkumný záměr MSM 604613730).
Literatura:
[1] Müller R.J., Schrader H., Profe J., Dresler K., Deckwer W.D.: Macromol. Rapid Commun. 26, 1400 (2005).
[2] Müller R.J., Kleeberg I., Deckwer W.D.: J. Biotechnol. 86, 87 (2001).
STUDY OF ADHESIVE PROPERTIES OF SELECTED POLYAMIDES MODIFIED BY PLASMA
Š. KURUC, I. NOVÁK*, I. CHODÁK
Polymer Institute of the Slovak Academy of Sciences, Dúbravská cesta 9, 842 36 Bratislava, Slovakia
E-mail: upolnovi/savba.sk
The polyamide 6 (Nylon 6, PA 6) foils were modified by dielectric
barrier discharge (DBD) plasma. A process of plasma modification was
done due to improving adhesion between PA 6 and more polar polymers.
The plasma treatment was made in oxygen, or nitrogen atmosphere. All
experiments were performed by atmospheric pressure (1 bar). Increasing
of adhesion was confirmed with two physical methods, i.e. peel testing,
and measurement of surface free energy by Surface Energy Evaluation
system (SEE system). The chemical modification of surface properties
was indicated by FTIR spectroscopy. ATR-FTIR spectroscopy was helpful
to show us creating of new source hydrophilicity on surface containing
polar functional groups like NH2- and -CO-NH2.
This contribution also shows the time behaviour of surface energy,
average peel strengths dependencies on the time of modification with
cold plasma in both atmosphere (N2 or O2). As a
result is significant increase of the surface energy and its polar
component of PA 6 modified by DBD plasma in oxygen or nitrogen plasma
at atmospheric pressure were confirmed. This increase significant even
of for short times of modification of polymer by plasma, and was
further increased for longer times of modification. The average peel
force of polyacrylate-bonded PA 6 modified by barrier discharge plasma
depends on the type of the plasma used.
The authors are grateful to the Slovak grant agency VEGA (grant No. 2/4042/04) for the financial support of this research.
SURFACE PROPERTIES OF POLYAMIDE 12 MODIFIED BY PLASMA AT ATMOSPHERIC PRESSURE
M. ŠTEVIAR, I. NOVÁK, I. CHODÁK
Polymer Institute, Slovak Academy of Sciences, Dúbravská cesta 9, 842 36 Bratislava, Slovak Republic
E - mail: upolnovi/savba.sk
To obtain a higher strength of adhesive joints of
polyamide 12 (PA 12) to more polar polymers, it is necessary to
increase its surface energy by specific modification methods. The most
advanced method of polymer surface modification is based on
modification by radio-frequency (RF) and barrier discharge plasma. The
surface of PA 12 was modified at various times in O2 or N2
plasma at atmospheric pressure (barrier discharge) or in air at reduced
pressure (RF-discharge). The adhesive properties of plasma modified PA
12 were evaluated by measurements of contact angles of the testing
liquids on the surface of polymer, peeling of adhesive joints, using
XPS and ATR-FTIR spectroscopy. The surface properties, i.e. the surface
energy and its polar component of plasma modified PA 12 were
determined. Evaluation of obtained results led to the conclusion that
the significant increase in hydrophilicity as well as in surface
energy, mainly in its polar component, in comparison with unmodified
polymer was reached after modification of PA 12 in N2 atmosphere.
The authors are grateful to the Slovak grant agency VEGA (grant No. 2/4042/04) for the financial support of this research.
COMPARISON OF SURFACE PROPERTIES OF SELECTED POLYPROPYLENES: INFLUENCE OF MALEATION
J. JŮZAa, I. NOVÁKb, Š. FLORIÁNb, V. POLLÁKb
a) Institute of Macromolecular Chemistry, Academy of Sciences of the
Czech Republic, Heyrovského nám. 2, CZ-162 06 Praha 6, Czech Republic (juza/imc.cas.cz; https://www.imc.cas.cz/ )
b) Polymer Institute, Slovak Academy of Sciences, Dúbravská cesta 9, 842 36 Bratislava, Slovak Republic; (upolnovi/savba.sk; http://www.polymer.sav.sk/ )
Introduction
In the frame of study of the influence of chemical modification on
the surface and adhesion of polypropylenes, surface tensions of several
polypropylenes were investigated.
Measurement methods
Surface tensions of melted copolymer isotactic polypropylene (iPP)
grafted with maleic anhydride (MA) were measured by the pendant drop
method [1] using an apparatus built according the description in
literature [2]. The samples were degassed either by melting at 200 °C
in an evacuated vessel for approx. 15 min before measurement, or only
at room temperature for several hours. The way of preparation showed no
significant influence on results.
Measurements were performed in argon. The image was read using Elvia
OS-458 CCD camera with Anaret 4,5/105 mm objective on tube of length
ca. 25 cm using Matrox IP-8 frame grabber with resolution 768x576
pixels at 256 bright levels. The profile was primarily detected by
tracing using Pavlidis algorithm [3, 4] and further refined with
maximal brightness gradient and partial edge smoothing [5]. The profile
was then processed using the multiple selected planes method [6] using
the interpolation equations that analogically to Misak's procedure [7]
derived previously [8].
To evaluate surface tension using this method, densities were
measured by time-consuming mercury dilatometry. Since the measurements
described were not the focus of research, we compared the primary
quantity, the ratio of surface tension to density; to obtain surface
tension, the density of a single sample, Mosten PH4, was used.
The surface tensions of the iPP films grafted with MA were measured
at laboratory temperature using an Amplival Pol [Zeiss, Germany]
goniometer, where the size of the drop spread with a micropipette
(Wheaton, USA) was 3 μl. Measurements were evaluated using 8 test
liquids with different polarity. The relationship of Owens, Wendt and
Fowkes adapted by Kinloch [9 -11] was used to compute the surface
energy and its components.
The adhesion joint strength in peeling was measured by the peel test
at the angle 90° on an Instron 4301 dynamometer [Instron, United
Kingdom] with an additional peeling circle device enabling keeping a
constant angle of 90° of peeling of grafted iPP film from the substrate.
Compared polypropylenes
|
Polypropylene
|
Mn
|
Mw
|
MA content
|
Melting point
|
|
|
|
|
% wt
|
°C
|
|
Tatren HPF 194
|
|
127400
|
0
|
158
|
|
Tatren non stabil.
|
69200
|
126300
|
0
|
156
|
|
IPP grafted in quasi solid state
|
62900
|
122600
|
0.4
|
163
|
|
Orevac 17832
|
28600
|
56500
|
0.8
|
144
|
|
Epolene G3003 mal.
|
27200
|
52000
|
1.2
|
158
|
|
|
|
Mosten 52422 a
|
|
370000
|
|
172
|
|
Mosten 52522 a
|
|
333000
|
|
172
|
|
Mosten 53692 a
|
38000
|
280000 (350000)
|
|
172
|
|
Mosten PH1 a
|
|
650000
|
|
172
|
|
Mosten PH4 a
|
|
350000
|
|
172
|
|
Mosten PH6 a
|
55000
|
300000
|
|
173
|
a Mostens contain 95-97 % of isotactic PP
Results processing
A typical feature of viscous and not completely homogeneous
materials like the polymers measured is that a substantial amount of
scanned drop profiles differ measurably and non-negligibly from the
theoretical shape [12] described by the Laplace equation. The measure
of that difference is a variance or sampling deviation of the values
determined for the same drop using respective selected planes. It
appears that the usual range of the deviations is different for each
sample. The maximal deviation from a model (theoretical) shape that
allows the drop to involve its result to further processing should be
selected the least possible, but leaving still enough data for further
processing. This maximal allowable deviation can be chosen either for
each temperature individually, when the criterion is more strict in the
middle of measurability range with a sufficient amount of quality drops
than near to the melting point or at the limit of the sample stability
range, or each sample individually but fixed for the whole temperature
range, or common for the set of samples compared.
We decided to apply the common criterion for the sample set. The
condition at which the variance of final results is acceptable and the
whole measured temperature range remains covered was the availability
of at least 3 selected planes and maximal sampling deviation on the
same drop of 0.2 mN/m.
For comparison, the selection was supplemented by results of
measurements of Mosten polypropylenes, where the primary results were
narrowed by the same criteria as the samples investigated now.
Data measured and found in literature
The following data were obtained for respective samples using the pendant drop method
|
Polypropylene
|
Measured range
|
γ at 200 °C
|
dγ /dT
|
106 γ/ρ (200 °C)
|
106d(γ/ρ) /dT
|
|
|
°C
|
mN.m-1
|
mN.m-1K-1
|
m3s-2
|
m3s-2K-1
|
|
Tatren HPF 194
|
180-243
|
19.4
|
-0.043
|
25.8
|
-0.038
|
|
Tatren non stabil.
|
180-240
|
19.2
|
-0.065
|
25.6
|
-0.068
|
|
iPP grafted
|
170-200
|
19.0
|
-0.052
|
25.4
|
-0.050
|
|
Orevac 17832
|
190-220
|
19.6
|
-0.12
|
26.1
|
-0.15
|
|
Epolene G003 mal.
|
180-230
|
19.4
|
-0.044
|
25.9
|
-0.039
|
|
|
|
Mosten 52422
|
200-240
|
19.5
|
-0.062
|
26.0
|
-0.064
|
|
Mosten 52522
|
200-250
|
17.1
|
-0.005
|
22.8
|
+0.012
|
|
Mosten 53692
|
180-210
|
19.2
|
-0.055
|
25.5
|
-0.052
|
|
Mosten PH1
|
190-240
|
19.8
|
-0.044
|
26.3
|
-0.040
|
|
Mosten PH4
|
180-230
|
19.0
|
-0.083
|
25.3
|
-0.092
|
|
Mosten PH6
|
180-230
|
19.6
|
-0.037
|
26.2
|
-0.028
|
|
Comparison with some other polypropylenes published in literature
|
|
|
|
|
|
Method
|
Author
|
|
PP atactic
|
120-190
|
19.7
|
-0.058
|
SB
|
Oda [14,15]
|
|
PP isotactic
|
190-230
|
19.7
|
-0.058
|
SB
|
Oda [14,15]
|
|
PP atactic
|
140-190
|
19.3
|
-0.056
|
PD
|
Roe [16]
|
|
Epolene Mn=3000
|
165-220
|
21.1
|
-0.040
|
RT
|
Schonhorn [17]
|
|
PP XAV 10A FOB
|
160-235
|
19.9
|
-0.0538
|
PD
|
Pötschke [18]
|
(PD = pendant drop method, SB = sessile bubble , RT = ring tensiometry)
Evaluation of surface energies of maleated iPP at room temperature
confirmed a significant increase in its values in comparison with
non-grafted iPP, mainly due to an increase in the polar surface energy
component. On the other hand, the comparison shows that the differences
in measured surface tensions of the melted samples with different
degrees of grafting with maleic anhydride were smaller than those
between respective non-grafted polypropylenes measured previously or
described in literature.
Whereas the surface energy could increase with a higher maleation
degree in the solid state, the materials that would increase the
surface energy of the system move in the melt into the bulk and lower
the total energy of the system. Hence, surface tension does not
increase.
Strengths of adhesive joints of iPP to poly(vinyl acetate) at
peeling significantly increased after grafting with maleic anhydride.
The work was supported by grant 106/06/0729 of Grant Agency of the
Czech Republic (a) and grant No 2/4024/04 of Slovak Grant Agency (VEGA)
(b).
- [1] Andreas, J. M.; Hauser, E. A.; Tucker, W.B. J Phys Chem 42, 1001 (1938).
- [2] Wu, S. J Colloid Interface Sci, 31, 153 (1969).
- [3] Pavlidis, T. in Algorithms for Graphics and Image Processing; Computer Sci. Press, Inc.: Rockville, (MD), USA, 1982; pp. 142‑148.
- [4] Hlaváč, V.;
Mařík, R.; Šára, R. Programmes and documentation of Department of
Control Engineering, Faculty of Electrical Engineering, Czech Technical
University: Prague, 1991.
- [5] Račinský, S. Ph.D. Thesis. J. Heyrovský Institute of Physical Chemistry and Electrochemistry, Czechoslovak Academy of Sciences: Praha, 1992.
- [6] Roe, R.J.; Bachetta, V. L.; Wong, P. M. G. J Phys Chem 71, 4190 (1967).
- [7] Misak, M. D. J Colloid Interface Sci 27, 141 (1968).
- [8] Jůza, J. Czech J Phys 47, 351 (1997).
[9] Novák I., Florián Š.: J. Mater. Sci. 39, 2033 (2004)
[10] Novák I., Pollák, V., Chodák I. Plasma Proces. Polyms. 3, 355-364, (2006)
[11] Novak I., Števiar M., Chodák, I. Monatsh Chem/Chem Monthly, June 2006, published online.
[12] Padday, J.F. in Surface and Colloid Science, Volume 1; Matijević, E., ,Ed.; Wiley-Interscience 1969; p. 103.
[13] Wu S.: J Macromol Sci, Part C 10,1‑73 (1974)
[14] Oda Y., Hata T.: Preprints, 267, 17th Annual Meeting of the High Polymer Societyof Japan (May 1968); cited from [13]
[15] Oda Y., Hata T.: Preprints, 6th Symposium on Adhesion and Adhesives, Osaka, Japan, June 1968, p.69; cited from [13]
[16] Roe R.J.: J Phys Chem 72, 2013 (1968)
[17] Schonhorn H.,Sharpe L.H.: J Polym Sci B 3, 235, (1965)
[18] Pötschke P., Pionteck J., Stutz H.: Polymer 43, 6965 (2002)
POVRCHOVÁ MODIFIKÁCIA TEXTILNÝCH MATERIÁLOV PLAZMOU
I. HUDEC*, Ľ. ČERNÁKOVÁ, M. JAŠŠO, J. ŠIROKÝ
Fakulta chemickej a potravinárskej technológie STU v Bratislave,
Ústav polymérnych materiálov, Oddelenie plastov a kaučuku, Radlinského
9, 812 37 Bratislava Slovenská republika, (ivan.hudec/stuba.sk)
Povrchová úprava textilných materiálov používaných v odevníctve,
resp. technických aplikáciách umožňuje modifikovať ich vlastnosti a
rozširuje spektrum aplikácií o nové oblasti. Jednou z možností
modifikácie povrchových vlastností textilných materiálov je aplikácia
plazmových technológií pracujúcich pri atmosferickom tlaku, ktoré
umožňujú rýchlu, ekonomicky a environmentálne výhodnú rovnomernú
povrchovú úpravu. Ich veľkou prednosťou je:
- modifikácia tenkých povrchových vrstiev v nanometrickej škále;
- suchý spôsob úpravy s malými nárokmi na spotrebu energie;
- zachovanie mechanických vlastností pôvodného materiálu.
Plazmu ako vysoko reaktívne prostredie pozostávajúce z elektrónov,
radikálov, kladne a záporne nabitých iónov, excitovaných molekúl a
elektromagnetického žiarenia je možné v oblasti modifikácie polymérov
využiť viacerými postupmi (obr. l).
 |
| funkcionalizácia |
aktivácia |
očkovanie |
depozícia |
| čistenie |
sieťovanie |
|
ultratenkých |
| leptanie |
|
|
filmov |
| Obr. 1 Možnosti použitia plazmy pri úprave polymérnych povrchov |
V rámci experimentov sa využilo viacero spôsobov modifikácie povrchu
textilných materiálov plazmou počnúc funkcionalizáciou, cez očkovanie
až po depozíciu ultratenkých filmov vytvorených plazmovou
polymerizáciou. Na aktiváciu textilných materiálov sa využil difúzny
koplanárny povrchový bariérový výboj (DCSBD) napájaný vysokofrekvenčným
striedavým napätím generujúci vrstvu plazmy na povrchu dielektrika.
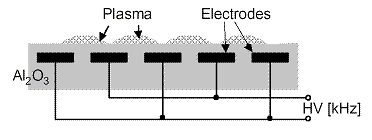
Schéma usporiadania elektród DCSBD výboja je na obr. 2.
 |
|
Obr. 2 Schéma usporiadania elektród difúzneho koplanárneho povrchového bariérového výboja (DCSBD) |
Výhodou uvedeného typu zariadenia je priamy kontakt textilného
materiálu s plazmou a relatívne vysoká energia plazmy umožňujúca krátky
čas kontaktu vedúci k homogénnej povrchovej úprave.
Povrchová modifikácia sa uskutočnila na netkaných polypropylénových textilných materiáloch (50g/m2), polypropylénových tkaninách (211g/m2), polyamidových tkaninách (86g/m2), polyesterových tkaninách (105g/m2), ako aj polyesterových kordových textíliách používaných ako výstužný materiál v gumárenskom priemysle.
V prednáške budú prezentované výsledky hydrofilizácie povrchu
viacerých typov textilných materiálov plazmou generovanou v rôznych
plynoch a za rôznych podmienok, hodnotenie stability hydrofilných
úprav, ako aj ovplyvnenie mechanických vlastností povrchovou
modifikáciou. V prípade polypropylénových netkaných aj tkaných textílií
sa na plazmou aktivovaný povrch naväzoval chitozán s cieľom prípravy
textílií s antibakteriálnymi vlastnosťami.
Povrchová modifikácia polyesterových kordov plazmou bola smerovaná
hlavne na zvýšenie ich adhézie ku gumárenskej nánosovej zmesi, pričom
sa študoval vplyv technologických podmienok modifikácie, typu plynu,
ako aj možnosti uplatnenia sa plazmovej polymerizácie na povrchovú
úpravu výstužných materiálov.
Poďakovanie
Autori práce vyjadrujú poďakovanie grantovej agentúre APVV za
finančnú podporu výskumu v uvedenej oblasti (projekt APVT-99-035004).
synthesis of poly(3-ethynylthiophene) with Homogeneous and Heterogeneous Rh Catalysts; polymer characterization
G. DVOŘÁKOVÁa, J. SVOBODAa, O. POŠTAa, J. SEDLÁČEKa, J. VOHLÍDALa
a Department of Physical and Macromolecular Chemistry,
Laboratory of Specialty Polymers, Faculty of Science, Charles
University, Albertov 2030, CZ-128 40, Prague 2, Czech Republic
(gita.dvorakova/post.cz, js/vivien.natur.cuni.cz)
Introduction
Substituted polyacetylenes attract attention due to their potential
applicability in electronics, optoelectronics and other fields [1,2].
Functional properties of these polymers can be tuned through both main
chain configuration structure (cis-trans and head-to tail isomerism)
and the choice of pendant groups. Polyacetylenes are mostly prepared by
chain polymerization of corresponding substituted acetylenes induced
with metathesis or insertion catalysts. As insertion catalysts various
Rh(I)(cyclodiene) complexes are used because of their high activity,
selectivity and precise control of configuration structure of resulting
polymers. Rh complexes provide highly stereoregular (cis-transoid,
head-to-tail) polyacetylenes molecules of which easily adopt the
helical conformation in the solid state. Moreover, these catalysts (i)
show unusually high tolerance to reaction surroundings, (ii) polymerize
acetylenes with a great variety of functional groups and (iii) can be
anchored on various inorganic and polymeric supports to give effective
heterogeneous catalysts [3].
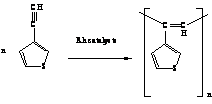 Since
thiophene units containing polymers are of great interests in the field
of conjugated polymers we have focused on polymerization of
3-ethynylthiophene (EtTh) (SCHEME 1). Results achieved with various
homogeneous and heterogeneous Rh catalysts including poly(EtTh)
characterization are reported in this contribution.
Since
thiophene units containing polymers are of great interests in the field
of conjugated polymers we have focused on polymerization of
3-ethynylthiophene (EtTh) (SCHEME 1). Results achieved with various
homogeneous and heterogeneous Rh catalysts including poly(EtTh)
characterization are reported in this contribution.
Results and Discussion
Polymerization
Complexes [Rh(COD)acac], [Rh(NBD)acac] and [Rh(NBD)Cl]2
(COD = cycloocta-1,5-diene, NBD = norbornadiene, acac =
acetylacetonato) were applied as homogeneous catalysts. Heterogeneous
catalysts tested were prepared as follows (i) [Rh(COD)Cl]2/PBI (Rh loading 3 wt.%) by direct anchoring [Rh(COD)Cl]2 from THF solution on porous polybenzimidazole (PBI) [4], (ii) [Rh(COD)Cl]2/APTMS/MCM41 (Rh loading 1 wt.%) by anchoring [Rh(COD)Cl]2 from CH2Cl2 solution on mesoporous molecular sieves MCM-41 surface modified with NH2(CH2)3Si(OCH3)3
linker (APTMS) [5]. Results of EtTh polymerization induced with these
catalysts are summarized in TAB. 1, examples of the time course of
reaction (monitored by SEC) are given in FIG. 1. Both mononuclear
[Rh(cyclodiene)acac] complexes provide good or moderate yield of
poly(EtTh) of weight-average molecular weight, Mw = 1.104 - 5.104 (Mw/Mn = 1.9 -2.7). Lower yield achieved in THF as compared to CH2Cl2
solvent may reflect a slight competitive inhibition of polymerization
by THF which is able to coordinate to Rh polymerization centres
(contrary to CH2Cl2). In both solvent tested
higher yield and particularly higher molecular weight of poly(EtTh) was
achieved with [Rh(NBD)acac] as compared to its COD containing
counterpart (Tab. 1, FIG. 1). However, the molecular weight of polymer
is not effectively controlled by the monomer to Rh mole ratio. Better
control on poly(EtTh) molecular weight was achieved with [Rh(NBD)Cl]2 applied in combination with triethylamine (Et3N) (Et3N/Rh = 10/1, Et3N promotes dissociation of [Rh(NBD)Cl]2
into mononuclear particles active in polymerization [1,3]).
Polymerization of EtTh performed with heterogeneous catalysts aimed at
preparation of high purity poly(EtTh) showed moderate polymerization
activity of [Rh(COD)Cl]2/PBI. Poly(EtTh) yields up to 25% (Mw ~ 1.104, Mw/Mn
~ 2.5) were achieved under conditions optimized so far. The fact that
TON value strongly increases with increasing initial monomer
concentration indicates that the diffusion of EtTh molecules into pores
of catalyst can play an important role in the overall polymerization.
Contrary to [Rh(COD)Cl]2/PBI, [Rh(COD)Cl]2/APTMS/MCM41,
which is known as highly active in phenylacetylene (PhA) polymerization
[5], was inactive in polymerization of EtTh.
Tab. 1 - Yield (Y) and weight-average molecular weight, Mw,
of poly(EtTh) and TON values achieved in EtTh polymerization with
homogeneous and heterogeneous Rh catalysts, room temperature, 5 h.
|
Catalyst
|
[EtTh]
M
|
[Rh]
mM
|
Solvent
|
Y
in %
|
10-3 Mw
|
TON
|
|
[Rh(NBD)acac]
|
0.6
|
6
|
THF
|
61
|
32
|
61
|
|
[Rh(NBD)acac]
|
0.6
|
6
|
CH2Cl2
|
69
|
45
|
69
|
|
[Rh(NBD)acac]
|
0.6
|
2
|
CH2Cl2
|
26
|
54
|
130
|
|
[Rh(COD)acac]
|
0.6
|
6
|
THF
|
30
|
14
|
30
|
|
[Rh(COD)acac]
|
0.6
|
6
|
CH2Cl2
|
61
|
19
|
67
|
|
[Rh(COD)acac]
|
3.0
|
6
|
CH2Cl2
|
57
|
14
|
285
|
|
[Rh(NBD)Cl]2/Et3Na)
|
0.6
|
6
|
CH2Cl2
|
100
|
20
|
100
|
|
[Rh(NBD)Cl]2/Et3Na)
|
0.6
|
2
|
CH2Cl2
|
98
|
46
|
300
|
|
[Rh(NBD)Cl]2/Et3Na)
|
0.6
|
1
|
CH2Cl2
|
38
|
86
|
228
|
|
[Rh(COD)Cl]2/
APTMS/MCM41
|
0.6
|
1.5
|
CH2Cl2
|
0c)
|
-
|
0
|
|
[Rh(COD)Cl]2/PBI
|
0.6
|
6
|
THF
|
8.5b)
|
10b)
|
8.5b)
|
|
[Rh(COD)Cl]2/PBI
|
0.6
|
24
|
THF
|
20b)
|
8.5b)
|
5b)
|
|
[Rh(COD)Cl]2/PBI
|
3.0
|
6
|
THF
|
25b)
|
13b)
|
125b)
|
[Rh(NBD)Cl]2/Et3N mole ratio = 1/10 b) reaction time 24 h
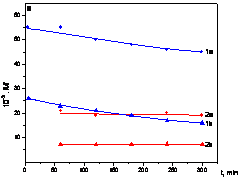
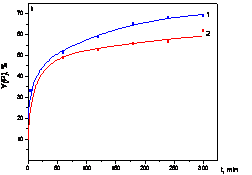
Fig. 1 - Yield (Y) of poly(EtTh) (I) and M
w and M
n values (II) vs. reaction time (t) for EtTh polymerization with [Rh(NBD)acac] (Y vs. t - curve 1, M
w vs. t - curve 1a, M
n vs. t curve 1b) and with [Rh(COD)acac] (Y vs. t - curve 2, M
w vs. t - curve 2a, M
n vs. t curve 2b). [Rh] = 6 mmol/l, [EtTh] = 0.6 mol/l, CH
2Cl
2, room temperature.
Polymer characterization
All prepared polymers are yellow-brown solids. Freshly prepared polymers are well soluble in THF, CH2Cl2, CHCl3
and toluene. Upon ageing in the solid state (room temperature, air,
time period of several months) the solubility has partly deteriorated.
Fig. 2 shows 1H NMR spectra of EtTh and representative
poly(EtTh) sample. Signal assignment was made on the basis of
3-ethylthiophene and poly(PhA) 1H NMR spectra [3, 4]. FIG.
2 shows (i) the absence of signal of monomer ºCH hydrogen and (ii)
preservation of signals of thiophene-ring hydrogens in the spectrum of
poly(EtTh) that confirms polymerization proceeds in accordance with
Scheme 1. By analogy to assignment of signal in 1H NMR
spectra of poly(PhA) the signal at 5.97 ppm in poly(EtTh) spectrum can
be ascribed to vinylic hydrogen of cis-transoid sequences. Well
development of this signal testifies to high degree of cis-transoid
configuration of the poly(EtTh) main chains. Using relation (1) cis
double bonds content in poly(EtTh)s prepared was estimated to be 80 -
90 % (Acis represents area of cis vinylic hydrogens, Atot is the area of signals of all hydrogens)
%cis = 400 . Acis/Atot (1)
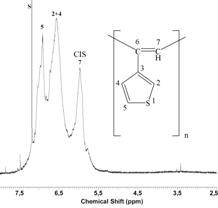
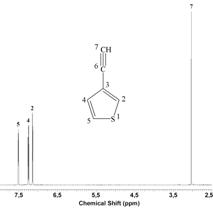
FIG. 2 - 1H NMR spectrum (in CDCl3) of EtTh and poly(EtTh) prepared with [Rh(NBD)acac].
Acknowledgement:
Financial support form the Grant Agency of the Czech Republic (project No. 203/05/2194) is gratefully acknowledged.
References
- M.C. Mayershofer; O. Nuyken J. Pol. Sci. A, Polym. Chem. 2005, 43, 5723.
- N.J. Long Angew. Chem. Int. Engl. 1995, 34, 41.
- J. Sedlacek; J. Vohlidal Collect. Czech. Chem. Commun. 2003, 68, 1745.
- J. Sedlacek; M. Pacovska; D. Redrova; H. Balcar; A. Biffis; B. Corain; J. Vohlidal Chem. Eur. J. 2002, 8, 366.
- H. Balcar; J. Cejka; J. Sedlacek; J. Svoboda, J. Zednik, Z. Bastl, V. Bosacek, J. Vohlidal, J. Mol. Catal. A-Chem. 2003, 203, 287.
PREPARATION OF POLYANILINES BY CATALYTIC POLYMERIZATION
M. BLÁHAa, M. ŽIGONb, J. VOHLÍDALb
aDept. of Physical and Macromolecular Chemistry, Faculty
of Science, Charles University, Albertov 2030, CZ-128 43 Praha 2, Czech
Republic
(Michal.Blaha/csop.cz; Vohlidal/natur.cuni.cz)
bLaboratory for Polymer Chemistry and Technology,
National Institute of Chemistry, Hajdrihova 19, POB 660, SI-1001
Ljubljana, Slovenia (Majda.Zigon/ki.si)
Oxidation system Fe3+/H2O2 easily
polymerizes aniline (ANI), 2-methoxy-ANI (OMA) and 2-chloro-ANI (CANI)
anilines giving high yields of corresponding polyanilines (PANIs) at
temperatures from 0 °C to 20 °C. The results obtained show that Fe3+ ions and HO·radicals
form a synergistic system for polymerization of ANIs, in which the ions
are key component in the first part and radicals in the second part of
connecting ANI unit into PANI chain. Quality of PANIs formed
catalytically is worse than that of corresponding PANIs prepared by
stoichiometric reactions, which should be ascribed to side reactions
induced by free radicals present in the reaction system.
Introduction
Polyaniline (PANI) is routinely produced by the stoichiometric
oxidative polymerization of aniline (ANI), which also gives a high
amount of waste side products1. Therefore, the development
of processes increasing purity of neat PANI and lowering the waste
production is highly desirable. This target can be potentially achieved
by using a suitable catalyst system.
ANI has already been polymerized with (i) Cu2+/O21 and Fe3+/ozone2 systems, which both show rather low polymerization activity, (ii) enzyme-based systems H2O2/peroxidase3, which is too expensive, and (iii) Fe3+/H2O2 system4, which seems to be the best one, and polymerizes ANI, 2-ethyl-ANI and 2-propyl-ANI4 to corresponding PANIs even at the Fe/ANI mole ratio 1:500.
The mechanism of this reaction has been proposed by Sun et al.4c,d who observed that, above 60 °C, H2O2 itself polymerizes PANI. Hence they proposed that, in the Fe3+/H2O2 system, Fe ions induce decomposition of H2O2 to HO·radicals,
which indeed polymerize ANI. However, there are still another possible
mechanisms of this reaction: (i) ANI might be polymerized with Fe3+ ions and the formed Fe2+ ions reoxidized by H2O2 (Scheme 1), or (ii) both reaction modes can take part in the overall reaction (see further).
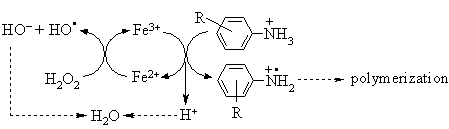
Scheme 1.Proposed catalytic cycle in the catalytic oxidative polymerization of ANIs with the system Fe3+/H2O2.
In the present paper we report on polymerization of ANI and
ring-substituted ANI derivatives: 2-chloro-ANI (CANI); and
2-methoxy-ANI (OMA) with the Fe3+/H2O2 system and discuss evidences pointing to complicated mechanism of this catalytic process.
Results and discussion
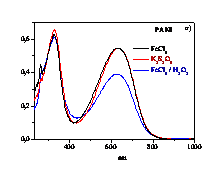
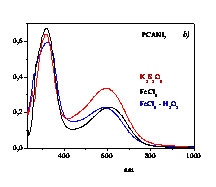
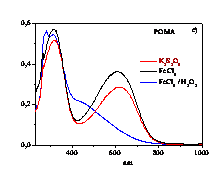 The system Fe3+/H2O2
applied in aqueous HCl (1 M) smoothly polymerizes all studied monomers
giving high yields (70 - 90 %) of corresponding polymers at room
temperature as well as at ca 0 - 5 °C. The UV/vis spectra (Fig. 1) show
that the system polymerizes ANI and CANI to the emeraldine forms,
which, however, show lower intensity of the Q-band (band at 600 nm
assigned to quinoid units) compared to that observed for PANIs prepared
stoichiometrically.
The system Fe3+/H2O2
applied in aqueous HCl (1 M) smoothly polymerizes all studied monomers
giving high yields (70 - 90 %) of corresponding polymers at room
temperature as well as at ca 0 - 5 °C. The UV/vis spectra (Fig. 1) show
that the system polymerizes ANI and CANI to the emeraldine forms,
which, however, show lower intensity of the Q-band (band at 600 nm
assigned to quinoid units) compared to that observed for PANIs prepared
stoichiometrically.
Figure 1 - UV/vis spectra of PANI, PCANI and POMA samples prepared by various procedures
Consistently, also conductivity of the polymers prepared with the Fe3+/H2O2 system reachs only 10 % to 50 % (s
» 0.5 S/cm) of that observed for stoichiometrically prepared polymers.
Characteristics of catalytically prepared poly(OMA) (POMA) are still
worse: (i) the Q-band is not developed in its UV/vis spectrum (Fig. 1c), and (ii) POMA does not become conductive upon doping with HCl (s » 10-11
S/cm). Surprisingly, no principle difference is seen between both FTIR
and FT Raman spectra of POMA samples prepared by the catalytic and
stoichiometric procedures, respectively. FTIR and FT Raman observations
indicate that PANIs prepared catalytically contain yet not specified
structure defects deteriorating their functional properties. The fact
that POMA electron-donating OCH3 pendent groups shows the structure dislocation much higher than PANI or PCANI indicates that side reactions of free HO·radicals cause these defects.
For ANI to be polymerized to the emeraldine form, 2.5 equivalents of the oxidant per one ANI equivalent are needed5. The [H2O2]/[ANI]
mole ratio equal to 1.25 (i.e., the same as if peroxodisulfate is used
as oxidant) was found to be sufficient to accomplish polymerization of
ANIs with FeCl3/H2O2 system. It means that radicals HO·formed as side product of reoxidation of Fe2+ ions (see Scheme 1) really participate as oxidant species in the overall polymerization process.
To get a better insight into the role of HO·radicals, we verified that H2O2 as well as CuCl2 by itself does not polymerize ANI at temperatures up to 20 °C and that CuCl2/H2O2 system, in which the decomposition of H2O2 to HO·radicals also takes place, is practically inactive at these temperatures. Thus, it can be concluded that FeCl3 is essential for the oxidation of anilinium ions (Scheme 1) while HO·radicals
act as oxidant in a further step of ANI polymerization, for example, in
the dehydrogenation of intermediate formed in combination of two
anilinium or anilinium and polyanilinium cation-radicals (Scheme 2).
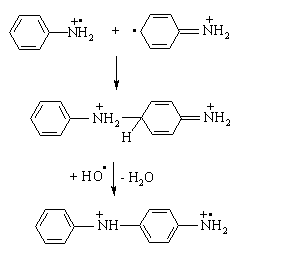
Scheme 2. Possible role of H2O2 in the catalytic polymerization of ANI with the system Fe3+/H2O2.
It is worth noting that, at temperatures about 0 °C, stoichiometric polymerization of ANI with FeCl3 gives only about 3 % yield (MW = 6 000) after 24 hours, whereas the catalytic polymerization with FeCl3/H2O2 gives the yield 84 % (MW 31 000). This suggests that low reactivity of Fe3+
ions in the dehydrogenation of the last-inbuilt monomeric units might
be the reason of the low to negligible polymerization activity of FeCl3 observed at lowered temperatures. Thus, it seems that Fe3+ ions and HO· radicals form a synergic system, in which iron ions are key component in the first stage and HO· radicals in the second stage of connecting an ANI unit into a PANI chain.
Acknowledgements
Financial supports of the Czech Science
Foundation (104/06/1087 and fellowship, M. Blaha, from project
203/03/H140) as well as travel supports of the Czech-Slovenian program
Kontakt (18/2005-06) and Ministry of Higher Education, Science and
Technology of the Republic of Slovenia and Slovenian Research Agency
(program P2-145) are greatly acknowledged.
References
- N. Toshima, S. Hara, Prog. Polym. Sci. 1995, 20, 155.
- a) Liu W., Kumar J., Tripathy S., Senecal J., Samuelson L., Synth. Met. 1999, 100, 71; b) Z. Jin, Y. X. Su, Y.X. Duan, Synth. Met. 2001, 122, 237; b) I. Y. Sakharov, A. C. Vorobiev, J. J. C. Leon, Enzyme Microb. Tech. 2003, 33, 661.
- Yan H., Kajita M., Toshima N. Macromol. Mater. Eng. 2002, 287, 503.
- a)D. K. Moon, K. Osakada, T. Maruyama, T. Yamamoto, Makromol. Chem. 1992, 193, 1723; b) D. K. Moon, K. Osakada, T. Maruyama, T. Yamamoto, Chem. Lett. 1991, 1633; c) Z. Sun, Y. Geng, J. Li, X. Jing, F. Wang, Synth. Met. 1997, 84, 99; d) Z. Sun, Y. Geng, J. Li, X. Jing, F. Wang, J. Appl. Polym. Sci. 1999, 72, 1077.
- J. Stejskal, P. Kratochvíl, A. D. Jenkins, Collect. Czech. Chem. Commun. 1995, 60, 1747.
- a) I. Mav, M. Žigon, J. Vohlídal, Macromol. Symp. 2004, 212, 307. b) I. Mav, M. Žigon, A. Šebenik, J. Vohlídal, J. Polym. Sci. Polym. Chem., 2000, 38, 3390.
Kinetic INVESTIGATION of olefin polymerization catalyzed by nickel-diimine complexes activated by non-MAO cocatalysts
J. Peleškaa, S. Hermanováa, J. MERNAb*
aInstitute of Material Science, Brno University of Technology, Purkyňova 118, CZ-612 00 Brno
bDepartment of Polymers, Institute of Chemical Technology Prague, Technická 5, CZ-166 28 Praha 6, jan.merna/vscht.cz
Kinetic investigation of propylene and hex-1-ene polymerization
initiated by
(N,N'-bis(2-t-butylphenyl)-1,8-naftalendiyl-1,4-diazabuta-1,3-diene)nickeldibromide
activated by methylaluminoxane (MAO), diethylaluminiumchloride (DEAC)
and ethylaluminiumdichloride (EADC) was performed. Hex-1-ene
polymerizations followed first order kinetics into high degree of
monomer conversion thus the apparent propagation constants could be
determined. The influence of polymerization temperature on
polymerization rate was examined in propylene polymerization and
apparent activation energy values were obtained. Further, the
dependence of molar masses and their distribution on reaction time and
temperature was studied. Correlation between molar mass and polymer
yield have shown the living character of all catalyst systems. The
differences of MAO, DEAC and EADC activation efficiency are discussed
in terms of their different Lewis acidity as well as differences in
counterion character. Finally, the influence of reaction condition and
catalyst composition on the microstructure of polymers was evaluated by
means of 1H NMR and DSC.
Phenoxy-imine catalyst for (di)olefin polymerisation
S. Hermanová, J. Cihlář, M. Kučera, J. Ševčík
Institute of Materials Chemistry, Faculty of Chemistry, Brno
University of Technology, Purkyňova 118, 612 00 Brno, Czech Republic
E-mail: hermanova-s/fch.vutbr.cz
Introduction
The diversity of possible structures of monomeric units in
macromolecules obtained by coordination polymerisation of dienes is an
interesting field for research. Next living polymerisation of dienes
permits the synthesis of block copolymers and chain-end modification.
Furthermore copolymerisation of alkenes with dienes would produce
polyalkenes with cyclic groups and (or) with pendant groups. This
contribution reports on the results of polymerisation of 1,5-hexadiene
with fluorinated (phenoxy-imine) titanium (FI Ti) complex activated by
methylaluminoxane (MAO) as cocatalyst. We focused on the conditions (T,
concentration of catalyst, ratio catalyst/cocatalyst) that maintain the
living way of polymerisation.
EXPERIMENTAL PART
All manipulations of air- and moisture-sensitive materials were
carried out under nitrogen atmosphere, using either a dual
vacuum/nitrogen line and standard Schlenk techniques or a high vacuum
line. Nitrogen (99.999 %, Siad) was purified by passing through 4A
molecular sieves and supported Cu columns. Toluene was obtained from
Lachema and was dried over sodium/benzophenone prior to use.
1,5-hexadiene (95%, Fluka) was dried over CaH2 and freshly distilled.
Bis[N-(3-tert-butylsalicylidene)-2,3,4,5,6-pentafluoroanilinato]titanium(IV)dichloride (1) was synthesized according to the previously described procedure 1.
Cocatalyst methylalumoxane (MAO) was purchased from Crompton GmbH
(10 % wt in toluene) and remaining trimethylaluminium was evaporated in
vacuo prior to use.
Polymerisations of 1,5-hexadiene were carried out in double-jacket
reactor (250 mL) with magnetic stirrer. Components were added as
follows; toluene, cocatalyst, monomer. The reactor was equilibrated at
appropriate temperature and after 20 minutes catalyst solution was
charged to start the polymerisation. The polymerisation was quenched by
adding 10 mL of ethanol, the polymer was precipitated in methanol/HCl,
filtered and then dried in vacuo to constant weight.
GPC analyses were recorded on SEC device using Polymer Laboratories
PLgel 10 mm MIXED-B, 300x7,5mm column, RI detector Ruby 05. Analyses
were conducted using THF as solvent at 25°C.
RESULTS AND DISCUSSION
Polymerisations of 1,5-hexadiene were performed in a toluene solution of 1/MAO.
The results are listed in Tab. I. Polydisperzities were unimodal and
reached the value <1.2 only at -10 °C. At temperature 0 °C the
broadening of molecular weight distribution was observed, indicating
that transfer (and/or termination) could take part (Figure 1). Lower
catalyst activity and degree of conversion observed at temperature -10
°C could be explained as the consequence of propagation rate reduction
as compared with the values measured at 0 °C.
The effect of dosing order was examined (Run 3). Surprisingly the result obtained by reverse introduction of 1
and MAO into the polymerisation mixture was better then the other runs.
The MAO is believed to behave also as the scavenger and hence it is
often added into the polymerisation solution before the catalysts.
The increase of catalyst amount at 10 mmol resulted in the decrease
of polymerisation activity (Run 5). The enhancement of polymerisation
activity using 5 mmol of 1 might be due to dilution effects favoring the dissociated ion pairs relative to their associated precursors.2
Table I Representative polymerisations of 1,5-Hexadienea
|
Run
|
n FI Ti [mmol]
|
Ratio
FI Ti / MAO
|
Ratio monomer / FI Ti
|
TP
[°C]
|
Yield
[g]
|
Mn b [kg.mol-1]
|
Mw/Mnb
|
Activityc
|
Conversion
|
|
1
|
5
|
1200
|
400
|
0
|
0.09
|
36
|
1.7
|
18000
|
0.55
|
|
2
|
5
|
1200
|
400
|
0
|
0.11
|
48
|
1.8
|
22000
|
0.65
|
|
3d
|
5
|
1200
|
400
|
0
|
0.15
|
67
|
1.6
|
30000
|
0.92
|
|
4
|
5
|
1200
|
400
|
-10
|
0.07
|
38
|
1.2
|
14000
|
0.44
|
|
5
|
10
|
300
|
200
|
0
|
0.16
|
41
|
1.5
|
16000
|
0.96
|
a Polymerisations were conducted in toluene (50 mL) using
2 mmol of hexadiene for each run. The polymerisation time was 60
minutes.
b Determined by GPC analysis using polystyrene standards and THF as the eluent
c[g of polymer/mol Ti]
d Reverse addition of catalytic components; MAO was introduced lastly
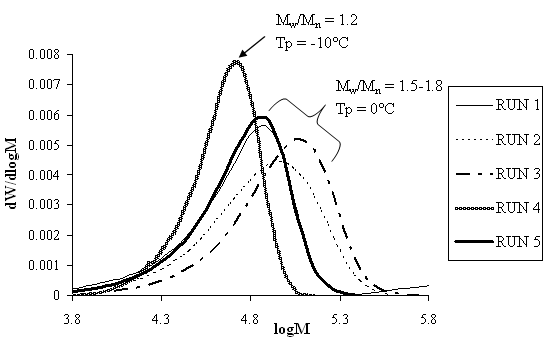
Figure 1 GPC chromatograms of prepared polymers
Conclusion
The aim of this work was to examined conditions of living polymerisations of 1,5-hexadiene catalyzed by bis[N-(3-tert-butylsalicylidene)-2,3,4,5,6-pentafluoroanilinato]titanium(IV)dichloride (1)
and MAO. Thepreliminary results have shown that the polymerisation
temperature should be decreased at -10 °C to avoid undesirable side
reaction such as transfer. The lowering of catalyst amount resulted in
the increase of polymerisation activity. The molecular weight increased
only slightly. The kinetics and mechanism of the polymerisation is
studied and the structure of products is revealed at the moment.
Acknowledgment
Acknowledgement is given to Project MSM0021630501
for financial support of this work.
REFERENCES
Fujita T., Mitani M.: J. Am. Chem. Soc. 124, 3327 (2002)
- Brintzinger H., H., Fischer D., Mülhaupt R., Rieger B., Waymount R., M.: Angew. Chem. Int. Ed. Engl. 34, 1143 (1995)
SEC/MALS STUDY OF POLY(3-ethynylthiophene) IN TOLUENE: MOLECULAR WEIGHT AND SIZE CHARACTERIZATION AND POLYMER DEGRADATION
D. RÉDROVÁ, J. SEDLÁČEK, J. ZEDNÍK, J. VOHLÍDAL
Department of Physical and Macromol. Chemistry,
Laboratory of Specialty Polymers, Faculty of Science, Charles
University, Albertov 2030, CZ-128 40, Prague 2, Czech Republic,
redrova/natur.cuni.cz
Introduction
Size Exclusion Chromatography (SEC) coupled with Multi Angle Light
Scattering (MALS) detector has been known as very powerful tool for
characterization of macromolecules in solution. Primarily it provides
the distributions of absolute values of molar mass (M) and radius of
gyration (Rg) and parameters of Rg vs. M dependence (scaling law 1).
Rg = k.Ms (1)
As we demonstrate in this contribution, SEC/MALS in combination with
SEC/calibration technique can be also sufficiently applied for
determination of constants of Kuhn-Mark-Houwink-Sakurada (KMHS)
equation (scaling law 2):
[h] = K.Ma (2)
Assuming that SEC separation is governed mainly by the hydrodynamic
volume of macromolecules that is proportional to the product [h].M,
where [h] is the limiting viscosity number, for each value of elution
volume relation (3) is obtained:
[h]1M1 = [h]2M2 (3)
where [h]1 and M1 is the limiting viscosity number and molar mass of the analysed polymer (M1 determined by SEC/MALS) and [h]2 and M2 corresponding values for the polymer standards used for the columns calibration for which KMHS constants (K2 and a2) are known. Combining (2) and (3) one derives Eq. (4):
log M1 = [1/(a1 +1)].log (K1/K2) + [(a2 + 1)/(a1 + 1)].log M2 (4)
KMHS constants for the analysed polymer are obtained from log M1 vs. log M2 plot.
In the case of polymers rapidly degrading in the solution SEC/MALS represents the only technique for reliable determination of Rg vs. M and [h] vs M dependences since it allows to determine at the same moment the sets of corresponding M, Rg
and [h] values. Moreover, SEC techniques is very powerful for
monitoring the course of polymer degradation and revealing the
character (random/nonrandom) of this process as we proved in the case
of several polymers of polyacetylene-type [1,2]. Coming from the Simha
equation that describes the kinetics of degradation in term of time
changes in degree of polymerization (DP) for each DP value [3], one can
derive for the case of random degradation following relations between
the degradation time (t) and number-average DP and weight-average DP, Pn and Pw respectively [4,5]:
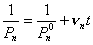 (5)
(5)
 (6)
(6)
where Iw is the index of polydispersity based on weight distribution of P, nn and nw the rate constants of degradation based on Pn and Pw
values, respectively. For the degradation to be of the purely random
character the experimental results must be represented by Eqs (5) and
(6) and determined nn and nw constants must be equal.
This contribution deals with the application of SEC/MALS to the
characterization of newly synthesized poly(3-ethynylthiophene) that
undergoes the autooxidative degradation in solution.
Results and discussion
Poly(3-ethynylthiophene) (P3ET) synthesized according to [6] was
dissolved in toluene (2 mg/ml) and the solution was stored in contact
to atmosphere at room temperature. At given times a withdrawn sample
was analysed by SEC/MALS apparatus using toluene as eluent to obtain
(i) distributions of absolute M and Rg values and
corresponding averages and (ii) apparent M distribution and averages
(relative to PS used for columns calibration). Representative samples
were simultaneously analysed by PS-calibrated SEC apparatus using THF
as eluent.
Results of degradation study are given in Tab. 1. Results evaluation
according to Eqs (5) and (6) shown in Fig. 1 reveals the random
character of degradation with values of rate constants: nn = 1,03.10-4 h-1 and nw = 1,02.10-4 h-1.
From Tab. 1 significant difference between the absolute and apparent M
averages is evident particularly for values obtained in toluene. It is
thus evident that P3ET macromolecules in toluene are more collapsed
than in THF solvent and that in both solvents they are far more
collapsed than PS ones of the same M. The last qualitative conclusion
is confirmed by values of scaling laws exponents (see Tab. 2) resulting
from evaluation of data obtained in the course of degradation according
to Eqs (1) and (4) (Fig. 2).
|

|
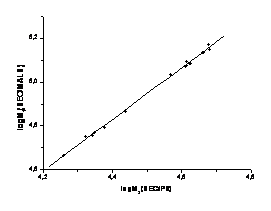
|
|
Fig. 1: 1/Pn vs. t (curve 1) and 1/Pw vs. T (curve 2) dependencies for degradation of P3ET in toluene at room temperature.
|
Fig. 2: Values of molar mass corresponding to the apex of SEC
peak in the course of P3ET degradation. SEC/MALS values plotted vs.
SEC/PS values according to Eq. 4.
|
Tab. 1. Molar mass and Rg averages determined by various
SEC techniques in the course of P3ET degradation in toluene solution
exposed to the atmosphere at room temperature.
|
t
in h
|
SEC/MALS
Toluene
|
SEC/PS calibration
Toluene
|
SEC/PS calibration
THF
|
|
|
10-3 Mw
|
10-3 Mn
|
Mw/Mn
|
(Rg)z
|
10-3 Mw
|
10-3 Mn
|
Mw/Mn
|
10-3 Mw
|
10-3 Mn
|
Mw/Mn
|
|
0,25
|
196
|
88
|
2,23
|
11,8
|
52
|
27
|
1,89
|
954
|
41
|
2,33
|
|
1,0
|
188
|
95
|
1,97
|
11,8
|
51
|
28
|
1,86
|
-
|
-
|
-
|
|
1,5
|
180
|
104
|
1,73
|
11,6
|
50
|
27
|
1,84
|
-
|
-
|
-
|
|
3,0
|
140
|
62
|
2,26
|
10,2
|
40
|
23
|
1,74
|
-
|
-
|
-
|
|
5,0
|
160
|
69
|
2,31
|
11,1
|
44
|
24
|
1,87
|
-
|
-
|
-
|
|
5,75
|
151
|
63
|
2,40
|
10,9
|
41
|
21
|
1,98
|
-
|
-
|
-
|
|
6,5
|
153
|
73
|
2,09
|
10,6
|
43
|
24
|
1,80
|
-
|
-
|
-
|
|
22
|
74
|
32
|
2,31
|
-
|
26
|
15
|
1,71
|
39
|
21
|
1,85
|
|
24
|
70
|
37
|
1,91
|
-
|
25
|
15
|
1,72
|
-
|
-
|
-
|
|
26,5
|
67
|
40
|
1,66
|
-
|
25
|
15
|
1,68
|
37
|
20
|
1,83
|
|
28
|
68
|
39
|
1,75
|
-
|
23
|
14
|
1,68
|
-
|
-
|
-
|
|
48
|
52
|
31
|
1,69
|
-
|
20
|
12
|
1,64
|
28
|
17
|
1,69
|
|
72
|
41
|
24
|
1,70
|
-
|
16
|
10
|
1,54
|
21
|
13
|
1,66
|
|
98
|
29
|
17
|
1,71
|
-
|
12
|
8,6
|
1,43
|
-
|
-
|
-
|
|
101
|
32
|
21
|
1,54
|
-
|
12
|
8,2
|
1,45
|
-
|
-
|
-
|
|
195
|
18
|
10
|
1,76
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
Tab. 2: Constants of scaling laws for P3ET and PS at room temperature
|
Polymer
|
Rg = k.Ms
|
[h] = K.Ma
|
|
Solvent
|
k in nm
|
s
|
Solvent
|
K in ml/g
|
a
|
|
P3ET
|
toluene
|
0,437
|
0,27
|
toluene
|
0,033
|
0,47
|
|
PS
|
toluene
|
0,0157
|
0,57
|
toluene
|
0,0093
|
0,73
|
|
PS
|
THF
|
0,0245
|
0,546
|
THF
|
0,0012
|
0,72
|
REFERENCES
- Sedláček J., Pacovská M., Vohlídal J., Grubišic-Gallot Z., Žigon M.: Macromol. Chem. Phys. 1995, 196, 1705.
- Rédrová D., Sedláček J., Žigon M., Vohlídal J.: Collect. Czech. Chem. Commun. 2005, 70, 1787.
- Simha R.: J. Appl. Phys. 1941, 12, 569.
- Vohlídal J., Rédrová D., Pacovská M., Sedláček J.: Collect. Czech. Chem. Commun. 1993, 58, 2651.
- Vohlídal J.: Macromol. Rapid. Commun. 1994, 15, 765.
- Dvořáková G., Svoboda J., Pošta O., Sedláček J., Vohlídal J.: Synthesis of Poly(3-ethynylthiophene) with Homogeneous and Heterogeneous Rh Catalysts. Poster at this Conference
PHOSPHORIC ACID-ACRYLATE MONOMERS FOR ADHESIVE DENTAL POLYMERS; POLYMERIZATION BEHAVIOR
J. PAVLINECa , N. MOSZNERb
aPolymer Institute, Slovak Academy of Sciences, Dúbravská cesta 9, SK-842 36 Bratislava, Slovak Republic ( upolpav/savba.sk)
bIvoclar Vivadent AG, Bendererstrasse 2, FL-9494 Schaan, Liechtenstein
Enamel-dentin adhesives are used to achieve a strong bond between a
filling composite and dental hard tissues (dentin and enamel). The
currently used self-etching enamel-dentin adhesives contain as adhesive
monomers particularly strongly acidic monomers, such as
methacrylic-group-containing phosphoric acids, which modify the enamel
and dentin surface and mediate the formation of a strong bond to the
composite filling material. In addition, cross-linking dimethacrylates
are used in order to improve the polymerisation reactivity of the
adhesive.[1] In self-etching adhesives, water is preferred
as a solvent. Thus, especially in the case of single-bottle adhesives,
the methacrylates undergo hydrolysis, which changes the chemical
composition of the adhesive and also impairs its performance. In this
respect, new phosphonic acids [2,3] and bis(acrylamide)s,
which feature an enhanced hydrolytic stability, can be used to
substitute commonly used methacrylates in self-etching enamel-dentin
adhesives. Recently, we were able to synthesise new steric hindered
a-methylsubstituted acrylate- containing dimethyl phosphonate groups.[4]
In this poster the homopolymerisation of the exceptionally
hydrolytically stable monomer 1 (Scheme 1), are described. Furthermore,
the copolymerisation of 1 with MMA, the polymerisable phosphonic acid 2
and the hydrolytically stable cross-linking bis(acrylamide) 3 were
investigated.
The polymerization behavior of bulky substituted polar
(meth)acrylates have not been still completely understood. In this
respect the present poster reports about the polymerization activity of
2,4,6-trimethylphenyl 2-[4-(dihydroxyphosphoryl)-2-oxa-butyl] acrylate
(monomer 1), carried out largely in THF and in temperature range of 55
-75 °C. The phosphoryl acid group makes the monomer molecule bipolar
and the polymerization alike of the other highly polar water- soluble
monomers depends on the ionization degree, and interactions with the
surrounding molecules.
The unusual polymerization rate-monomer concentration dependence
with monomer intensity exponent less than 1 (0.5) was assumed to be the
result of an increase in the propagation rate constant with THF monomer
dilution. The reaction order even more declined from 1 in the polar
EtOH-H2O polymerization medium.
More effective initiation with DBPO peroxide initiator compared with
AIBN was observed (at the same temperature). Moreover the reaction
order 0.36 instead of usual 0.5 points on the another reaction course
deviations from the conventional polymerization rate relationships.
Nevertheless the initial polymerization rates in dependence on
temperature gave straight lines in the Arrhenius coordinates with
slopes 81 kJ/mol, for both AIBN and DBPO initiated polymerization. This
value range well with Ea detected for another (meth)acrylate esters
polymerization.
The copolymerization kinetic data demonstrates also the solvent
effect, mutually on copolymer composition and copolymerization rate.
The local co-monomer composition around the propagating polymer chain
being different from the bulk may explain the changes in copolymer
composition by altering a solvent quality for copolymerization feed.
The water-soluble, hydrolytic stable monomers of this type are used
in dental filling composites to achieve a strong bond to the tooth
substances.
Scheme 1: Monomer 1

Figure 1. The initial polymerisation rates in dependence of the
reciprocal polymerisation temperature. The semi-logarithmic plot for
the polymerisation of 1 (1.0 mol/L) in THF with AIBN or DBPO (0.025 mol/L).
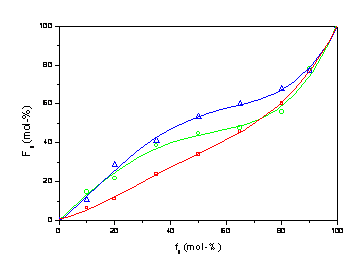
Figure 2.Copolymerization curves: Copolymer composition F1, 1 (mol-%) units in copolymer in dependence of f1, 1 (mol-%) in the monomer feed. Monomer combinations:(i/1)1,1 with 2 in THF; (i/2) O, 1 with 2 in EtOH-H2O (1:1 vol/vol); (ii) ê, 1 with MMA in THF
Scheme 2. Monomer 2 and monomer 3
References
[1] N. Moszner, Macromol. Symp. 2004, 217, 63.
[2] N. Moszner, F. Zeuner, J. Angermann, U.K. Fischer, V. Rheinberger, Macromol. Chem. Phys. 1999, 200, 1062.
[3] N. Moszner, F. Zeuner, S. Pfeiffer, I. Schurte, V. Rheinberger, M. Drache, Macromol. Mater. Eng. 2001, 286, 225.
[4] F. Zeuner, S. Quint, F. Geipel, N. Moszner, Synth. Commun. 2004, 34, 767.
Polypropylene/Boehmite nanocomposite fibres, spinning and mechanical properties
M. Hricová, A. Marcinčin
Slovak University of Technology in Bratislava, FCHPT, IPM,
Department of Fibres and Textile Chemistry, Radlinského 9, 812 37
Bratislava, SK, e-mail: marcela.hricova/stuba.sk
Development of the polypropylene (PP) based nano-composites,
containing layered silicates or other inorganic nanofillers is focused
mainly on improvement of the mechanical properties, enhancement of
electrical conductivity and barrier properties, thermal stability and
lowering of combustibility of polymers [1-4].
The idea to improve the mechanical properties (tenacity, modulus) of
polypropylene fibres by spinning of fibre forming polypropylene
nanocomposite consist in favourable arrangement of the structure
elements of oriented fibres resulting from: a) suppression of melt
elasticity in spinning, to obtain higher structure uniformity of fibres
(at low concentration of nanofiller), b) positive effect of nanofiller
on size of crystallites (nucleating effect), to obtain the fine
supermolecular structure of oriented fibres, c) reinforcing effect of
oriented nanoparticles in polymer matrix of fibres, d) creating three
dimensional net of nanoparticles in fibre matrix and e) creating
specific conditions in forming of fibres, to obtain higher orientation
of the amorphous region. It can be assumed that only lower
concentrations of nanofiller are needed (up to 1 wt %) for
above-mentioned control of the structure of PP fibres [5]. To improve
another properties of PP fibres such as barrier against UV radiation,
electrical conductivity and others, the higher concentrations of the
nanofiller are assumed (up to 5 wt%).
In this paper, the effect of spinning conditions on selected
mechanical properties of PP/Boehmite nanocomposite fibres is presented.
In experimental part the model PP/Boehmite nanocomposite fibres were
described using two-step process: preparation of PP nanofiller
concentrate and spinning of mixture of the PP and PP/nanofiller
concentrate. Two procedures of preparation of PP/Boehmite nanocomposite
fibres, method A and method B were used. Mechanical properties of
fibres are discussed with regard to their structural non-uniformity and
rheological properties of concentrated dispersions of nanofiller in PP.
The PP Moplen 500R and PP Moplen 561N (Basell Co) as well as PP HPF
(Slovnaft Co) were used in experimental work. Boehmite is marked in
this contribution as Boehmite S4. The original concentration of
Boehmite S4 in C1-C5 concentrates was 10 wt%. The concentrates were
pre-treated before spinning.
|
 1 1
|
 2 2
|
FIG. 1 Characteristic structure of the Boehmite S4 particle
FIG. 2 Method of preparation of polymer nanocomposite fibres
Rheological properties of the PP/Boehmite concentrates and spinning of PP nanocomposite fibres
The rheological properties of PP and PP/Boehmite
concentrates were measured using capillary extrusiometer Göttfert N
6967 with extruder f=20 mm at 280°C. Both Newton and Oswald de Waele
laws were used for determination of the basic rheological parameters:
apparent viscosity h=t/γ and power law exponent n (t = k.γn),
which characterize the non-Newtonian behaviour of the polymer melt, t -
shear stress, γ˙ - shear rate, k - coefficient. Results are given on
the Fig. 3 and in the Table 1.
FIG. 3 Dependence of the viscosity of polymer melts on shear rate at 280°C, for PP and concentrates C1-C4.
Table 1 Power law exponent n and viscosity h for PP and PP/Boehmite concentrates at 280°C.
|
Sample
|
n
|
h [Pa.s]
|
|
|
g=100 s-1
|
g=500 s-1
|
|
PP M 500R*
|
0,44
|
165
|
67
|
|
|
PP HPF**
|
0,42
|
284
|
111
|
|
|
PP M 561N
|
0,44
|
317
|
130
|
|
C1
|
0,41
|
235
|
90
|
|
|
C2
|
0,45
|
199
|
83
|
|
|
C3
|
0,46
|
273
|
115
|
|
|
C4
|
0,46
|
260
|
109
|
|
* T=250°C, ** T=275°C
The dependences on the Fig. 3 and results in the Table 1 reveal
that PP HPF exhibits a high deviation from the Newtonian flow (low
power law exponent n), which does not change significantly for
PP/Boehmite nanocomposite C1. Pre-treated C2-C4 concentrates exhibit
lower deviation from the Newtonian behaviour. The processing viscosity
of PP/Boehmite concentrates is lower compared to viscosity of powder
polymer PP HPF. The similar or lower viscosity of C1-C4 concentrates
relative to basic PP M 561N is convenient for mixing of PP and
concentrates before spinning [6].
Mechanical properties and non-uniformity of PP/Boehmite nanocomposite fibres
Instron type 3343 was used for evaluation of the mechanical
properties, tenacity, elongation and Young modulus of the PP/Boehmite
nanocomposite fibres (according to ISO 2062:1993). The variation
coefficients of the tenacity T, elongation at break E and Young modulus
YM were used as measure of the internal (structural) unevenness of the
PP/Boehmite fibres.
Table 2 Fineness Tdt, tenacity T, elongation E and Young
modulus YM of the PP M 561N/Boehmite S4 nanocomposite fibres, prepared
by method A as well as their coefficients of variation.
|
Concen
-trates
|
Nanoffiler
in fibres [%]
|
Tdt [dtex]
|
T [cN/dtex]
|
CVT [%]
|
E
[%]
|
CVE [%]
|
YM [cN/dtex]
|
CVYM [%]
|
|
-
|
-
|
98,4
|
5,56
|
1,6
|
25,4
|
10,0
|
62,4
|
4,4
|
|
C1
|
1,0
|
98,1
|
4,87
|
1,7
|
24,3
|
8,1
|
52,1
|
6,5
|
|
C2
|
1,0
|
98,8
|
5,10
|
1,8
|
26,5
|
9,1
|
54,3
|
5,1
|
|
C3
|
1,0
|
98,7
|
5,24
|
1,1
|
25,1
|
7,4
|
56,1
|
5,3
|
|
C4
|
1,0
|
98,3
|
4,24
|
4,3
|
33,8
|
22,1
|
49,7
|
5,8
|
The tenacity and Young modulus of fibres prepared by method A in
general are slightly lower than that for unmodified PP fibres (Table
2). In dependence on pre-treatment procedures, the tenacity of fibres
increases with decrease of non-uniformity of fibres (CV). The similar
correlation was found also for fibres prepared by method B (Table 3).
The lower non-uniformity, the higher tenacity and Young modulus of
fibres were found. The mechanical properties of PP nanocomposite fibres
prepared by method B (Table 3) reveal that concentrations of inorganic
nanofiller in PP under 1,0 wt% are more convenient. Already, at 0,05 -
0,1 wt% of nanofiller concentrations, the higher tenacity and Young
modulus of PP nanocomposite fibres were found out. Besides, the higher
content of nanofiller provides the higher deformability of composite
fibres. Elongation of fibres increases in this case (Table 3).
Table 3 Fineness Tdt, tenacity T, elongation E and Young
modulus YM of the PP M 561N/Boehmite S4 nanocomposite fibres prepared
by method B and their coefficients of variation.
|
Concen
-trate
|
Nanoffiler
in fibres [%]
|
Tdt
[dtex]
|
T
[cN/dtex]
|
CVT
[%]
|
E
[%]
|
CVE
[%]
|
YM
[cN/dtex]
|
CVYM
[%]
|
|
-
|
-
|
91,0
|
7,2
|
5,0
|
21,9
|
3,1
|
60,6
|
4,7
|
C5
|
0,02%
|
86,0
|
7,4
|
4,1
|
21,4
|
3,9
|
72,5
|
6,5
|
|
0,05%
|
90,0
|
7,5
|
2,8
|
20,6
|
4,8
|
71,8
|
3,3
|
|
0,1%
|
89,0
|
7,6
|
3,2
|
20,5
|
3,3
|
71,3
|
3,5
|
|
0,3%
|
98,0
|
6,9
|
3,8
|
22,9
|
6,6
|
65,5
|
5,4
|
|
1%
|
105,0
|
5,7
|
4,6
|
23,4
|
13,9
|
60,0
|
4,9
|
|
3%
|
122,0
|
4,4
|
5,0
|
30,5
|
13,6
|
45,6
|
5,7
|
Conclusion
The PP/Boehmite nanocomposite fibres were
prepared using two procedures. The fibres exhibited improved basic
mechanical properties in comparison with unmodified PP fibres already
at low content of nanofiller (0,05 - 0,1 wt%). Formulation of the
PP/Boehmite composition and spinning conditions were found as key
factors to improve the mechanical properties of fibres.
This work was supported by Nanohybrid-project, which is
part-financed by the Sixth Framework Programme of European Commission
(NMP3-CT-2005-516972).
References
- Hasegawa N., Okamoto H., Kato M., Tsukigase A., Usuki A.:
Polyolefin-clay hybrids based on modified polyolefins and organophilic
clay. Macromolecular Materials and Engineering, 76, 2000 p. 280-281
- Nam P. H., Maiti P., Okamoto M., Kotaka T., Hasegawa N., Usuki
A.: A hierarchical structure and properties of intercalated
polypropylene clay nanocomposites. Polymer 42(23), 2001, p. 9633-9640
- Alexandre M., Dubois P.: Polymer-layered silicate nanocomposites:
preparation, properties and uses of a new class of materials. Material
Science and Engineering 28, 2000, p. 1-63
- Tang Y., Hu Y., Song L., Zong R., Gui Z., Chen Z., Fau W.: Polymer Degradation and stability 82, 2003, p. 127-131
- Mlynarcikova Y, Borsig E., Legen J., Marcincin A., Alexy P.:
Influence of the Composition of Polypropylene/Organoclay Nanocomposite
Fibers on their Tensile Strength, J. Macromol. Sci., Part A: Pure and
Appl. Chem. 42, 2005, pp. 543-554
- A. Marcinčin, M. Hricová, J. Lučivjanský: Influence of the
Rheological Properties of Pigment Concentrates on Unevenness of
Polypropylene Spun-dyed Fibres, Fibres &Textiles in Eastern Europe,
13, No. 1, 2005, pp. 16-19
BARRIER PROPERTIES OF POLYPROPYLENE FIBRES MODIFIED BY NANOTiO2
M. DULÍKOVÁa, A. UJHELYIOVÁa, A. MARCINČINa, O. BREJKAb
a Faculty of Chemical and Food Technology, Slovak
University of Technology, Department of Fibres and Textile Chemistry,
Institute of Polymer Materials, Radlinského 9, SK-812 37 Bratislava,
Slovak Republic (maria.dulikova/stuba.sk, http://www.chtf.stuba.sk)
bResearch Institute for Man-Made Fibres, Join-Stock Company, Štúrova 2, SK-059 21 SVIT, Slovak Republic (http://www.vuchv.sk)
Introduction
Polymer materials have been filled with several inorganic
nanoadditives in order to increase several properties like barrier
properties, mechanical properties, thermal stability etc. Recently,
considerable attention has been devoted to the barrier properties of
textiles designed for clothing as a protection against UV radiation. UV
radiation is an invisible component of sunlight, divided into UVA
radiation (wavelength 315-400nm), UVB radiation (wavelength 280-315 nm)
and UVC (wavelength 100-280 nm). UVA and UVB radiation penetrate the
skin and may result in some skin damage such as, sunburn, premature
ageing of the skin, even skin cancer, eye damages, etc [1].
The findings reported in the literature concerning the barrier
properties of fabrics in relation to UV radiation show that attention
has been focused on the physical aspect of barrier properties of
fabrics or yarns used for fabric production. A relatively weak emphasis
has been put on the effect of fibre-forming polymers on the UV
absorbing capacity [2].
There have been investigated numerous fabrics of cotton, wool, silk,
polyester, polyamide etc. for their protective properties against UV
radiation and its capacity for improvement by UV absorbers in the
literature. The UPF of the textile was investigated in term of
molecular and morphological structure of the fibre and the substances -
additives or textile processing products contained in it as well as its
structural features - porosity/number of holes, thickness, dimensional
stability, elastic properties, etc [3].
"UV - protect factor" (UPF) is a value for increasing the natural
protection period of human imparted by a consumer product which
protects the area of skin from direct irradiation. The natural
protection period for human skin (approximate safe period in the sun)
depends on skin type [1].
In this paper the method of measuring of barrier properties
(transmittance) against UV radiation of fibres modified by TiO2
is presented. UV transmittance by textiles is determined by the
transmittance through the spaces between the yarns as well as the
transmittance through the fiber, which means that it depends on the
type of fibre and the textile construction used [4]. Incorporating UV
absorbents such as TiO2 to the fiber can decrease UV
transmittance through textile material (fibers). Another advantage is
that it cannot be removed by washing and present better coloration.
Experimental
Barrier properties of PP fibres modified by TiO2 were
measured by spectrophotometer "Libra S12". Polypropylene - TG 920 and
nanoadditive - hombitec S-100 were used for preparation of the modified
polypropylene/TiO2 (PP/TiO2) fibres. The fibres contained 0.2 - 3.0 wt. % nanoTiO2.
Sample of fibres at the measurement was prepared as follows: fibre
was reeled on metallic small windows grate-shaped. Small windows have
had different parameters, distances between cuts have been 0.5, 0.75
and 1 mm. Transmittance of individual samples were measured in 10
positions for each fibre and it was statistically evaluated.
Consequently UPF, UVA and UVB were calculated by standard specification
STN EN 13758-1:2001 using the following equation:
 ,
,
Eλ - relative erythermal spectral effectiveness (W/m2.nm), Sλ - solar UVR spectral irradiance (Melbourne), Tλ - spectral transmittance of the sample (%), Δλ - bandwidth (nm), λ - wavelength (nm).
Table 1 Transmittance of PP fibres with content of 3.0 wt. % nanoTiO
2 for the statistical evaluation
|
Sample/Position
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
|
A
|
6.1
|
6.4
|
6.3
|
7.4
|
9.3
|
11.7
|
10.2
|
9.4
|
7.7
|
8.1
|
|
B
|
9.4
|
8.9
|
9.6
|
11.6
|
11.1
|
12.4
|
12.7
|
12.6
|
12.6
|
11.2
|
|
C
|
4.7
|
4.5
|
4.2
|
4.3
|
4.7
|
5.2
|
6.7
|
6.8
|
5.3
|
5.5
|
|
D
|
13.7
|
13.3
|
8.9
|
7.0
|
7.6
|
6.8
|
6.5
|
6.3
|
7.4
|
6.8
|
|
E
|
5.5
|
6.3
|
6.5
|
6.0
|
6.8
|
7.8
|
8.5
|
8.6
|
9.4
|
9.4
|
|
F
|
11.2
|
17.0
|
13.8
|
13.2
|
11.6
|
12.5
|
12.4
|
12.2
|
13.7
|
12.8
|
|
G
|
14.1
|
21.2
|
27.3
|
25.6
|
23.5
|
19.7
|
13.4
|
14.4
|
11.9
|
9.5
|
|
H
|
6.8
|
9.9
|
19.3
|
29.4
|
19.8
|
22.0
|
16.1
|
13.3
|
12.4
|
9.6
|
|
I
|
6.9
|
8.1
|
9.5
|
9.1
|
8.9
|
7.5
|
7.7
|
10.2
|
8.1
|
6.7
|
|
J
|
6.7
|
8.6
|
9.1
|
9.0
|
9.7
|
10.8
|
13.9
|
12.5
|
10.5
|
11.6
|
|
K
|
19.2
|
20.6
|
20.8
|
26.3
|
32.8
|
13.2
|
18.9
|
12.4
|
9.5
|
8.5
|
|
L
|
4.8
|
4.7
|
4.9
|
5.9
|
6.6
|
5.7
|
5.6
|
6.0
|
5.7
|
5.6
|
|
Source of variability
|
SS
|
Deviation
|
MS
|
F
|
Value P
|
Fcritical
|
|
Rows
|
2268.05
|
11
|
206.19
|
15.73
|
2.64E-17
|
1.89
|
|
Columns
|
214.14
|
9
|
23.79
|
1.82
|
0.08
|
1.98
|
|
Mistake
|
1297.74
|
99
|
13.11
|
|
|
|
|
|
|
|
|
|
|
|
|
Sum
|
3779.93
|
119
|
|
|
|
|
Table 2 Barrier properties of PP fibre and PP fibres modified by nanoTiO2 - UPF, UVA, UVB and UVC
|
Sample
|
Content of TiO2, %
|
UPF
|
UVA
|
UVB
|
UVC
|
|
1
|
-
|
4.8
|
0.21
|
0.21
|
0.20
|
|
2
|
0.2
|
5.2
|
0.22
|
0.21
|
0.21
|
|
3
|
0.4
|
5.3
|
0.21
|
0.20
|
0.20
|
|
4
|
0.6
|
6.2
|
0.17
|
0.16
|
0.16
|
|
5
|
0.8
|
4.9
|
0.21
|
0.21
|
0.21
|
|
6
|
1.0
|
5.7
|
0.20
|
0.19
|
0.19
|
|
7
|
1.2
|
6.0
|
0.18
|
0.18
|
0.18
|
|
8
|
2.0
|
6.8
|
0.18
|
0.17
|
0.17
|
|
9
|
3.0
|
8.5
|
0.13
|
0.13
|
0.13
|
Results and discussion
The modification of the method at the measurement of UV radiation
transmittance using spectrophotometer was focused on evaluation of the
influence of reeling of the fibre on the small window and position of
small window in spectrophotometer. The influence was evaluated by
transmittance at ten different reeling of the fibre and ten positions
of small windows in spectrophotometer (Table 1). We observed from
obtained results and statistical evaluation that position of small
window in spectrophotometer has negligible effect on the transmittance
of fibres. However, individual reeling has expressive effect what
relate with evenness of linear density or diameter of fibres. It is
necessary to bear in mind the linear density, evenness and structure of
textile.
Following the barrier properties of PP fibre and PP fibres modified by different content of TiO2 were evaluated by UPF, UVA, UVB and UVC values (Table 2). UPF increases with the increasing content of nanoTiO2 and on the other side UVA, UVB and UVC decrease. Modification of PP fibres by nanoTiO2
up to 3.0 wt. % has positive effect on barrier properties but this
modification does not provide materials with values as presented in the
standards.
Acknowledgements
Support of the National Grant Agency of Slovakia APVT - Grant No 20-011404 is appreciated.
References
1. UV STANDARD, 801.
2. Joanna Alvarez, B.L.-S., Examination of the absorption properties
of various fibres in relation to UV radiation. AUTEX Research Journal,
2003. 3(2).
3. Reinert G., H.R., Schmidt E., Fuso F., Sonnenschutzeigenschaften
textiler Flächen und deren Verbesserung. Textilveredlung 31, 1996.
11/12.
4. Hanke D., H.K., Altmeyer P., Dermatological Clinic of the
University Bochum, Böhringer B., Schindler G., Schön U., Akzo Nobel
Fibres, Wuppertal, Klotz M. L., Technical College,
Mönchengladbach/Germany, UV protection by textiles. Chemical Fibres
International, 1997. 47.
GRAFTING OF ISOTACTIC POLYPROPYLENE WITH ITACONIC ANHYDRIDE BY REACTIVE EXTRUSION
F. KUČERA a
aFaculty of Chemistry, Brno University of Technology, Purkyňova 118, CZ-612 00 Brno, Czech Republic (kucera-f/fch.vutbr.cz)
Introduction
Polypropylene (PP) is one of the currently most produced and used
polymers, with world consumption expected 32,9 billion tones in 2006.
Pure polypropylene is sufficient for many applications, however
utilization of PP in other applications, such as composites, needs
improvement of some of its properties for better adhesion, miscibility,
dyeability etc. Chemical modification of common polymers is a way of
formation of new material without searching new monomers for
polymerization. Industrial and scientific interest gained in recent
years grafting of polar monomers onto PP chain [1, 2, 3, 4, 5], most of
modification reactions target the change of PP hydrophobia. Reaction of
PP with maleic anhydride (MA) is recently the best known modification
process [6, 7, 8], which is generally used for improvement of
polypropylene adhesion to metals, glass fibres or to other polymers
such as nylon or polyethyleneterephthalate. Grafting reaction by
reactive extrusion can be used for grafting of MA of other polar
compounds onto PP chain in a melt state of polymer and in presence of
free radical initiator. Polypropylene functionalized by MA is now
available on market and is also widely used. Possibility of grafting
other polar substances, such as other carboxylic derivates, vinyl or
acrylic molecules, were studied [9, 10, 11]. Itaconic anhydride is
monomer used for grafting to PP backbone in this work.
Experimental
Reactive extrusion was carried out in twin screw extruder type DSE 25 (Brabender OHG, Duisburg, Germany) at 200 °C and 20 rpm.
Melt Flow Index (MFI) was measured using MFI tester CEAST6542/000
(CEAST S.p.A., Torino, Italy) at temperature 230 °C and load 2,16 kg
according to ISO 1133 and ASTM D 1238.
FT-IR Spectroscopy was used to determination of content of grafted
IAH. Spectra were taken on FT-IR spectrometer Nicolet Impact 400 D
(Thermometric, USA). Characteristic peak for vibrations of methylene
groups of PP occurred at 1167 cm-1 and characteristic peak for vibrations of C=O groups of anhydride occurred at 1771 cm‑1.
Integral intensities of characteristic peaks were used for quantitative
determination of efficiency of grafting. Samples of grafted PP were
measured as thin films, which were prepared by hot pressing at 205 °C
for 20 minutes.
Reaction mixture of polypropylene powder with deposited additives on
its surface was prepared by mixing additives with PP powder. Calculated
amounts of chemicals were mixed using glass stick with the PP powder in
the acetone solution (150 ml of acetone used) for 5 minutes. Prepared
suspension was then evaporated in the vacuum to obtain dry reaction
mixture. Extruder was set up to the speed of 20 rpm and all heating
zones were set to the temperature of 200 °C. PP was dosed into the
input of the extruder and passed through machine. Head of the extruder
produced 2 strings, which were water-cooled and cut into small
granules.
Results and discussion
Experimental part was focused to study of three major influences on
grafting reactions: type and concentration of stabilization,
concentration of initiator and concentration of grafting agent.
The PP powder H4m in reaction system PP/Luperox 101/IAH as grafting
agent was stabilized using K10P of concentration 0,42 mmole/kg PP.
Obtained yields of grafting reaction with IAH depending on peroxide
concentration are presented in Fig. 1 Efficiency of grafting (a)
depends strongly on the peroxide concentration, with increasing
concentration of initiator, part of IAH grafted onto PP chain increase
in reason of increasing concentration of macroradicals.
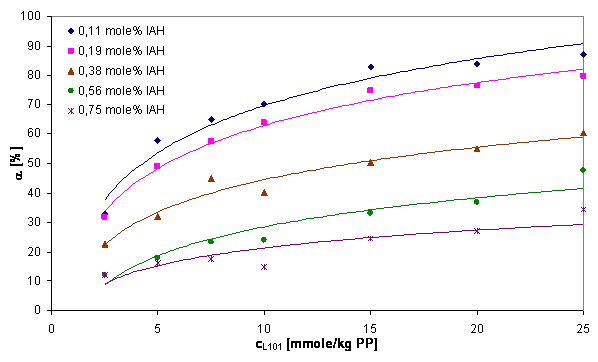
Fig. 1 Dependence of efficiency of grafting a on Luperox 101 concentration. Concentration of stabilizer Kingnox 10 P was 0,42 mmole/kg PP. Concentration of IAH ranging from 0,11 mole % to 0,75 mole %.
Maximal value of grafted itaconic anhydride on PP chain was
converting to 90 %, observed at peroxide concentration of 25 mmole/kg
PP and 0,11 mole % of IAH. But also concentrations 15 and 20 mmole/kg
PP and 0,11 mole % of IAH reached grafting yields over 80 %. High
yields of grafting could be theoretically acquired also with high
concentrations of IAH (0,38, 0,56 and 0,75 mole%), but to manage it,
higher than 25 mmole/kg PP of peroxide should be used.
Results of grafting reactions with IAH can be also summarized as dependence of c (IAH)PP on c (IAH)0
in Fig. 2 Results show that with increasing initial concentration of
IAH, concentration of grafted anhydride increase. Dependence reaches
maximal grafted concentration of IAH, which could be grafted onto PP
chain (around c (IAH)0=0,4 mole%). And afterwards
concentration of grafted agent decreases or stays the same. Adding
higher amount of IAH than 0,4 mole% seems to be inefficient, because
majority of this amount won't be possible to graft onto PP chain.
Low amounts of IAH grafted on PP, of concentrations higher than c (IAH)0=0,4 mole%, are probably caused by the chemical structure of IAH radical.
IAH radical is of low reactivity, caused by delocalization of
electrons in IAH structure. With increasing initial concentration of
IAH probability of forming of IAH radical increase, because probability
of attack of IAH by primary radical grows.
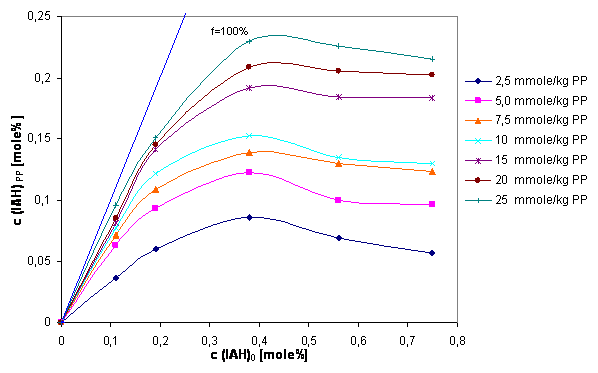
Fig. 2 Dependence of concentration of IAH grafted onto PP versus initial concentration of IAH. CK10P=0,42 mmole/kg of PP. CL101 was from 2,5 mmole/kg PP to 25 mmole/kg PP.
Conclusions
Primary stabilizer Kingnox 10P was selected as the most effective
stabilization for grafting reactions on PP. Luperox 101 was chosen for
initiation of free radical grafting reactions on PP chain. With
increasing concentration of Luperox 101, yield of grafting reaction
increased, but Mw of modified PP decreased in case of b-scission. Mw of grafted PP decreased with decreasing concentration of itaconic anhydride, used as grafting agents.
References
[1] Al-Malaika S.: Reactive modifiers for polymers. Blackie A@P, New York 1997
[2] Yazdani-Pedram M., Vega H., Retuert J., Quijada R.: Pol. Eng. Sci., 43(4), 2003, 1789
[3] Yazdani-Pedram M., Quijada R., López-Manchado M. A.: Macromol. Mater. Eng.,1, 2003, 288
[4] Sain M. M., Kokta B. V.: J. of Reinforced Plastics and Composites, 13, 1994, 657
[5] Sain M. M., Kokta B. V.: Polym.-Plast. Technol. Eng., 33 (1), (1994), 89-104
[6] Dean S., Jinghui Y., Zhanhai Y., Yong W., Hongliang H., Giovanna Costa: Polymer 42, 2001, 5549
[7] S. M. Ghahari, H. Nazokdast: Intern. Pol. Processing XVIII (2003) 3, Hanser Publishers, Munich.
[8] Zámorský Z.: Karboxylace polypropylénu v pevné fázi. Plasty a kaučuk, 30 (9), 1993, 72
[9] Yazdani-Pedram M., Vega H., Quijada R.: Polymer 42, 2001, 4751
[10] Yazdani-Pedram M., Vega H., Quijada R.: Macromol. Chem. and Phys.36, 1998. 2495
[11] Pesetskii S. S., Jurkowski B., Makarenko O. A.: J. of Appl. Pol. Sci., 86, 2002, 64
Acknowledgement
This work was supported by GAČR, grant 203/03/D165 and by Czech Ministry of Education, grant MSM 0021630501.
COMPARISON OF UHMWPE STRUCTURE IN THE NEAT FORM AND IN HIGHLY OXIDIZED COMPONENTS OF TOTAL JOINT REPLACEMENTS
E. PAVLOVAa, H. SYNKOVÁa, M. ŠLOUF a, D.POKORNÝb, A. SOSNAb
aInstitute of Macromolecular
Chemistry, Academy of Sciences of the Czech Republic, Heyrovskeho nam.
2, 162 06 Praha 6, Czech Republic
bOrthopaedics Clinic, Hospital Motol, V Úvalu 84, 156 06 Praha 6, Czech Republic
Total joint replacements, which are used in surgery, consist of two
main parts: the first is metallic and the second is made of ultrahigh
molecular weight polyethylene (UHMWPE). The polymeric component may
undergo long-term oxidative degradation. This leads to complex changes
of polymer structure and material embrittlement, associated with a
subsurface defect called white band (Fig 1, 2).
|

|
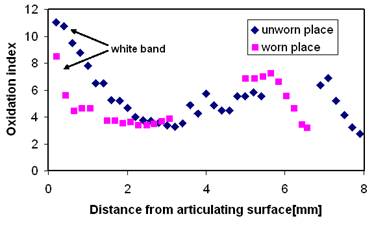
|
|
Fig. 1. Light micrograph showing 60 mm cross-section through the damaged UHMWPE cup. Inner surface of the cup is on the left.
|
Fig. 2. Oxidation index of the damaged UHMWPE cup after
revision, plotted as a function of distance from the inner surface of
the cup. The oxidation index gives the relative amount of C=O bonds in
the sample.
|
In this study, we compared structural changes and extent of
oxidation in three kinds of samples: (i) neat UHMWPE (Chirulen 1020),
(ii) gamma-irradiated UHMWPE prepared in laboratory and (iii)
commercial UHMWPE cups from damaged, joint replacement after revision.
Light microscopy (LM) and transmission electron microscopy (TEM) were
used for structural characterization, scanning electron microscopy
(SEM) with microanalysis (EDS) was employed in investigation of
articulating/inner surfaces of damaged UHMWPE cups. Infrared
spectroscopy (FTIR) gave the quantitative data for evaluation of
oxidative degradation.
Various kinds and levels of surface damage were found for UHMWPE's
from different manufacturers. Extent of oxidative degradation
(determined by FT-IR and microstructure analysis) depended not only on
the material and its origin, but also on the distance from the
articulating surface.
Acknowledgement: grant GAČR 106/04/1118.
VLIV OZAŘOVÁNÍ A TEPELNÉ ÚPRAVY NA ROZPUSTNOST UHMWPE
H. SYNKOVÁ*, M. ŠLOUF, Z. STARÝ, J. BALDRIAN
Institute of Macromolecular Chemistry, Academy of Sciences of the
Czech Republic, Heyrovsky sq. 2, CZ-162 06 Praha 6, Czech Republic (*synkova/imc.cas.cz)
V moderní medicíně se již řadu let používá vysokomolekulární
polyethylen (UHMWPE) jako jedna z hlavních komponent totálních
endoprotéz (TJR). Přestože má tento polymer vynikající mechanické,
frikční a otěrové vlastnosti, jeho životnost je limitována. Za hlavní
příčinu selhání TJR jsou považovány submikronové částice UHMWPE, které
vznikají otěrem polymerní komponenty o kovovou hlavici.
Odolnost UHMWPE vůči otěru lze zvýšit sesíťováním polymerních
řetězců ionizujícím zářením. V našem případě jsme použili dva druhy
záření: urychlené elektrony (dávka 0, 50 a 100 kGy) a γ-záření (dávka
0, 25, 50 a 100 kGy). Ozářené vzorky byly následně tepelně upraveny při
teplotě vyšší než je teplota tání UHMWPE (tzv. remelting = RM) za
účelem deaktivace zbylých volných radikálů, jejichž přítomnost vede k
oxidativní degradaci materiálu. Sesíťováním a následnou tepelnou
úpravou dojde k výrazným změnám ve struktuře krystalické a amorfní
části UHMWPE. Strukturu UHMWPE lze zkoumat pomocí celé řady
mikroskopických, spektrálních, difrakčních i termických metod.
Na výsledný stupeň sesíťování UHMWPE lze usuzovat např. ze
stanovení nerozpustného podílu. Na základě předběžných experimentů bylo
zjištěno, že doba dostačující k dosažení rovnovážného nerozpustného
podílu při botnání ve vroucím xylenu je 8 hodin. Ze získaných výsledků
vyplývá, že ozářením UHMWPE dojde k výraznému zvýšení obsahu
nerozpustného podílu a to bez ohledu na typ použitého záření (Obr. 1 a
2). Z hlediska dosažení vyššího obsahu nerozpustného podílu a tedy i
vyššího stupně sesíťování je vhodné vystavit UHMWPE účinkům urychlených
elektronů, tedy záření o vyšší energii. V tomto případě nebyly
pozorovány změny rozpustnosti v závislosti na dávce ozáření (Obr. 1).
Naproti tomu v případě γ-záření docházelo se zvyšující se dávkou
ozáření ke zvyšování obsahu nerozpustného podílu (Obr. 2). Z
předložených dat je dále patrné, že tepelná úprava ozářených vzorků
nemá výrazný vliv na rozpustnost UHMWPE.
Obrázek 1. Vliv ozáření urychlenými elektrony a následné tepelné úpravy na rozpustnost UHMWPE.
Obrázek 2. Vliv ozáření γ-zářením a následné tepelné úpravy na rozpustnost UHMWPE
Práce byla financována z grantu GAČR 106/04/1118 a podpořena Akademií Věd ČR (projekt AVOZ40500505).
SYNTHESIS AND PHOTOPHYSICAL PROPERTIES OF NOVEL CONJUGATED ACYCLIC ARYLAMINE DERIVATIVES
A. BÍLEŠOVÁ1, C. KÓSA1, J. MOSNÁČEK1, M. DANKO1, P. KASÁK2
1Polymer Institute Slovak Academy of Sciences, Dubravska cesta 9, 842 36 Bratislava, Slovak Republic
2Department of Organic Chemistry, Faculty of Natural
Sciences, Comenius University, Mlynska dolina, 842 15 Bratislava,
Slovak Republic
Presenting autor's e-mail: upolbila/savba.sk
Study of free nitroxyl radicals has a great importance for several
reason. They play an important role in the mechanism of light and
partially heat stabilization of polyolefins. In this case, stable free
nitroxyl radicals are formed from their precursors - parent amines
(HAS) during stabilization [1]. The free nitroxyl radical can influence
the photochemical and photophysical processes due to its paramagnetic
effect [2].
In recent years, the free nitroxyl radicals is coupled with the new
polymerization method, living free radical polymerization (LFRP). This
technique offers the control of molar mass and molar mass distribution
(polydispersity) in the radical polymerization and the possibility of
preparation of block copolymers with required design [3, 4].
We intend to study the influence of electron-donating substituents
in a structure of amine on the photophysical properties, because the
structure of the amines and electrone properties of substituents have
an influence at the oxidation of amines to nitroxyl radical, as well as
at their disproportionation during oxidation reaction.
New acyclic aryl amine derivatieves
(1,2-dimethyl-1-phenylpropyl)(4-methoxyphenyl)amine and
(1,2-dimethyl-1-phenylpropyl)(4-methylphenyl)amine have been
synthesized and characterized. Their photophysical properties have been
investigated in non-polar and polar solutions and in PS and PMMA
polymer matrices. Influence of the substituent in para-position of
aniline group as well as the environment polarity to the absorption,
emission spectra and fluorescence quantum yield has been observed.
[1] E. N. Step, N. J. Turro, P. P. Klemchuk and M. E. Gande, Angew. Macromol. Chem. 232 (1995) 65.
[2] V. A. Kuzmin, A. S. Tatikolov, Chem. Phys. Lett. 51 (1977) 45.
[3] C. J. Hawker, Acc. Chem. Res.,30 (1997) 373.
[4] K. Matyjaszewski, In Controlled Radical Polymerization, K.
Matyjaszewski, Ed., American Chemical Society: Washington, DC, 685
(1998) 2.
FAST THERMORESPONSIVE ORGANIC-INORGANIC POLY(N-ISOPROPYLACRYLAMIDE) BASED HYBRID SYSTEMS
B. STRACHOTOVÁ, L. MATĚJKA,A. STRACHOTA, M. ŠLOUF, J. PLEŠTIL
Institute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic,
Heyrovského nám. 2, CZ-162 06 Praha 6, Czech Republic (beata/imc.cas.cz)
Poly (N-isoprpoylacrylamide) hydrogels are a subject of lively
scientific interest for a long time, due to the "volume phase
transition", which they show under certain conditions. Linear
poly(NIPA) undergoes a discontinuous phase separation and precipitates
from its aqueous solution at a temperature above 32 °C, which is the
lower critical solution temperature (LCST) of poly(NIPA) in water.
Cross-linked poly(NIPA) gels swollen with water undergo a discontinuous
volume transition if the temperature is raised above the LCST of linear
poly(NIPA). This feature makes poly(NIPA)-based systems a very
attractive subject for investigation, with many possible applications
like drug delivery systems, actuators, molecular valves, the
immobilization of enzymes, nano-valves, artificial muscles etc. Simple
poly(NIPA) bulk hydrogels show slow swelling-deswelling kinetics and
poor mechanical properties at high swelling degrees. However, a fast
response to environmental stimuli is crucial for the majority of
applications.
In this contribution we present the preparation of very fast responsive
organic-inorganic (O‑I) hybrid gels from N-isopropylacrylamide (NIPA),
N,N'-methylenebisacrylamide (BAA) and tetramethoxysilane (TMOS) by
simultaneous radical polymerization and hydrolytic polycondensation of
TMOS in aqueous solution (see Fig.1).
|
 |
|
Fig.1 Formation of poly (NIPA)-based O-I networks with silica structures dispersed in the organic matrix |
The gel syntheses were carried out at temperatures below the LCST of
poly(NIPA) in two steps. The first step was performed at +15 °C and during the second step the temperature was lowered to ‑18 °C.
The ice crystals, which grew during the second stage, served as the
pore-forming agent. Under appropriate synthesis conditions, an
open-celled pore structure is formed in the hydrogels and makes
possible a fast, convective, water transport inwards and outwards of
the gel, in contrast to slow water transport by diffusion in bulk gels.
The prepared hydrogels display not only a rapid deswelling, but in
contrast to most fast responsive poly(NIPA) gels described in
literature, they show also a very fast reswelling. The hard inorganic
domains play a very important role, as they stabilize the pore
structure of the O-I hydrogels and besides that they improve the gels
mechanical properties.
The authors thank the Grant Agency of the Czech Republic, Grant Nr. 203/05/2252 for the financial support of this work.
AMIDATION OF O-(CARBOXYMETHYLSTARCH): PREPARATION, CHARACTERISATION AND PROPERTIES
Z. JAKUBÍKOVÁ 1, I. SROKOVÁ 1, V. SASINKOVÁ 2 , D. MOŠKOVÁ 3, A. EBRINGEROVÁ2
1 Faculty of Industrial Technologies TnU of A. Dubček, I. Krasku 491/30, 020 01 Púchov, Slovak Republic
2 Center of Excellence of the Slovak
Academy of Sciences, Institute of Chemistry, Dúbravská cesta 9, 845 38
Bratislava, Slovak Republic
3 Institute of Polymers, Slovak Academy
of Sciences, Dúbravská cesta 9, 845 38 Bratislava, Slovak Republic,
jakubikova/fpt.tnuni.sk
Partially hydrophobized CMS derivatives represent important
biosurfactants [1, 2]. An interesting alternative to the conventional
hydrophobization methods, based on the esterification of carbohydrates
with acid halides, anhydrides or by transesterification represents the
amidation of CMS with alkyl amines [3]. Recently, a performing metod of
amidation of carboxymethylcellulose [3] and alginates has been reported
by xy et al., 1999 [5].
This contribution deals with partial hydrophobization of
O-(carboxymethyl)starch (CMS, DS = 0.3) with laurylamine. The reaction
was performed in methanol and 4-DMAP as catalyst at laboratory
temperature (20°C)
Various reaction conditions were studied in order to optimize the
preparation of water-soluble CMS derivate. The obtained derivatives
were characterized by FT-IR spectroscopy and the surface-active
properties were determined by surface tension, emulsifying efficiency
and foamability.
The results suggested the partially hydrophobized CMS derivatives
are potential amphiphilic biosurfactants, which might be exploited in
various technical applications.
Acknowledgements: This work was financially supported by the Slovak Grant Agency VEGA, project No. 2/6131/6 and No. 1/3161/06.
References
[1] Sroková, I., Miníková, S., Ebringerová, A., Sasinková, V., Heinze, Th., Tenside Surf. Det., 40 (2003) 73.
[2] Burczyk, B., Encyclopedia of Surface and Colloid Science, Marcel Dekker, ISBN: 0-8247-0859-8 (2002) 724.
[3] Charpentier, D., Mocanu, G., Carpov, A., Chapelle, S., Merle, L., Miller, G., Carbohydrate Polymers 33(1997) 177-186.
Ecological vulcanizing Systems based on thiophosphoryl sulFides
Z. Hrdlička, V. Holčák, A. Kuta
Department of Polymers, Institute of Chemical
Technology, Technická 5, Prague 6, 166 28, Czech Republic,
zdenek.hrdlicka/vscht.cz
A vulcanizing system is a necessary part of a rubber
compound. It joins the original linear rubber chains to a
three-dimensional network of the cured rubber. Elementary sulphur is
still the mostly used curing agent. A vulcanizing accelerator is an
important part of the curing system as well. It increases the rate of
vulcanization and also the crosslinking efficiency. Many kinds of
accelerators are being used today.
| Fig. 1: Tetramethylthiuram disulphide |
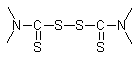 |
|
Tetraalkylthiuram disulphides (or polysulphides) are very fast
vulcanizing accelerators. They can also act as curing agents in
compounds without elementary sulphur and so sulphur-free vulcanization
can be carried out. However when heated, they can release secondary
amines which they are derived from. These amines further undergo
nitrosation to form carcinogenic nitrosamines.
One way how to eliminate the nitrosamine pollution
is to use vulcanizing accelerators that don't produce secondary amines.
This paper deals with ecologically harmless vulcanizing systems based
on thiophosphoryl derivatives (see Fig. 2). Commercially available
mixture of tetrasulphides (TDDP) was chosen. The system was used for
sulphur and sulphur-free vulcanization of natural (NR) and
styrene-butadiene (SBR) rubber. The aim was to find the optimal
composition of the system which could lead to the optimal course of
vulcanization and to a sufficient extent of crosslinking as well.
| Fig. 2: Bis(O,O'-diialkylthiophosphoryl) tetrasulphide |
 |
|
Curing characteristics of the rubber compounds were measured at 140 °C using Rubber Process Analyzer (RPA 2000, Alpha Technologies).
The results were compared to those obtained with a conventional curing
accelerator - tetramethylthiuram disulphide (TMTD). Mechanical
properties of the cured rubber haven't been determined but they could
be assumed from the extent of crosslinking (DM). Some other
substances had to be added to the system, viz.thiourea (TU),
N,N'-diphenylguanidine (DPG) or triethanolamine (TEA).
The results show that the optimal curing system for
NR contains TDDP 6 phr, DPG 2 phr and sulphur 1 phr. The optimal
formulation for SBR is TDDP 6 phr, TEA 2 phr and sulphur 1 phr.
However, a vulcanizing system is only one part of the rubber compound
formulation and the influence of other additives on the environment has
to be tested.
VYUŽITIE FLUORESCENCIE PRE ŠTÚDIUM ŠTRUKTÚRY ROZTOKOV VODOROZPUSTNÝCH POLYMÉROV V ZMESNOM ROZPÚŠŤADLE VODA -MONOMÉR
1Ústav polymérov, Slovenská akadémia vied, Dúbravská cesta 9, 842 36 Bratislava, Slovenská republika
2Medzinárodné laserové centrum, Ilkovičova 3, 821 19 Bratislava, Slovenská republika
E-mail: katarina.mikusova/savba.sk, igor.lacik/savba.sk
Vodorozpustné polyméry predstavujú skupinu makromolekulových látok
so špecifickými vlastnosťami určenými polárnymi alebo iónovými
skupinami v ich chemickej štruktúre, ktoré sa prejavujú rôznym typom
interakcií predovšetkým ak je voda použitá ako rozpúštadlo. Následne
vodorozpustné polyméry nachádzajú široké uplatnenie v rôznych
oblastiach, ako napríklad v ekológii ako flokulanty, superabsorbenty, v
kozmetike ako zahusťovače, prášky na pranie, v medicíne a farmakológií
ako dočasné membrány, kapsule, nosiče liečiv a ako priemyselné polyméry
v papierenskom a textilnom priemysle, atď. Napriek veľkému množstvu
aplikácii, mechanizmus radikálovej polymerizácie vodorozpustných
monomérov polymerizovaných vo vodnej fáze sa až do nedávna opieral o
výsledky získané spred 30 rokov. V posledných rokoch významný progres v
kinetike a mechanizme radikálovej polymerizácie vodorozpustných
monomérov polymerizovaných vo vodnej fáze bol dosiahnutý použitím
kombinácie polymerizácie iniciovanej pulzným laserom spojenej s
charakterizáciou molekulovej hmotnosti gélovou permeačnou
chromatografiou vo vodnej fáze. Príkladom je aktualizácia výsledkov
získaných zo štúdií kinetiky propagácie kyseliny metakrylovej
polymerizovanej vo vodnej fáze.[1]
Kvôli množstvu interakcií vo vodnej fáze bolo často diskutované
akým spôsobom sa mení štruktúra a dynamika polymerizujúceho roztoku
počas polymerizácie a či tieto interakcie významne ovplyvňujú kinetiku
polymerizácie. Tieto diskusie boli založené na roztokových
vlastnostiach vodorozpustných polymérov vo vodnej fáze, kde je voda
použitá ako rozpúšťadlo. V polymerizačnom roztoku sa rozpúšťadlo skladá
z vody a monoméru, čo samozrejme ovplyvňuje konformáciu polymérneho
klbka v porovnaní so situáciou, keď sa ako rozpúšťadlo použije voda.
Posun zloženia rozpúšťadla od čistej vody k čistému monoméru môže viesť
k výraznému ovplyvneniu štruktúry roztokov z pohľadu vytvárania
polárnych a nepolárnych domén, preferenčnej adsorbie jednej zložky
zmesného rozpúšťadla polymérnym klbkom a pod. Tieto zmeny je možné
zachytiť využitím napr. fluorescencie s vhodne zvolenou fluorescenčnou
značkou, čo je princípom aj nášho štúdia.
Tento príspevok hovorí o našej práci venovanej roztokom
neionizovanej kyseliny polymetakrylovej a kyseliny polyakrylovej o
rôznej koncentrácii pri rôznych pomeroch zmesi voda a príslušný
monomér. Predbežné fluorescenčné merania pri použitím pyrénu ako
fluorescenčnej značky ukázali, že charakteristiky emisného spektra
fluorescencie pyrénu, t.j. pomer I1/I3 a vytváranie exciméru, ako aj zhášanie emisie pyrénu v prítomnosti monoméru sú silne závislé od zloženia rozpúšťadla.
Poďakovanie
Táto práca bola podporovaná projektom APVV-51-037905 a VEGA grantom s číslom 2/6015/6.
Literatúra
[1] S. Beuermann, M. Buback, P. Hesse, I. Lacík. Macromolecules 39 (2006) 184.
MOŽNOSTI PŘÍPRAVY POLYESTERAMIDOVÝCH NANOKOMPOZITŮ
J. KREDATUSOVÁ, J. BROŽEK, J. SVOBODOVÁ
Ústav polymerů, Vysoká škola chemicko - technologická v Praze, Technická 5, Praha 6, ČR (kredatuj/vscht.cz)
Polyesteramidy (PEA) lze připravit aniontovou kopolymerací ε-kaprolaktonu (CLO)
a ε-kaprolaktamu (CLA). S rostoucím obsahem CLO jednotek ve struktuře
polyesteramidového řetězce se zvyšuje citlivost k biologické
rozložitelnosti, ale zároveň dochází ke zhoršení některých mechanických
vlastností - klesá modul pružnosti v tahu a pevnost. U jejich
nanokompozitů by bylo možno tyto vlastnosti zlepšit již při nízké
koncentraci plniva.
Nanokompozity představují poměrně novou skupinou materiálů, které se
obecně skládají z vrstevnatého silikátu dispergovaného jako ztužující
fáze v polymerní matrici. Připravují se třemi hlavními způsoby -
míšením v tavenině, polymerací in situ a exfoliovanou adsorpcí.
Pro přípravu polyesteramidových nanokompozitů jsme využili metodu exfoliované adsorpce a polymerace in situ, k jejich charakterizaci byly využity termické metody DSC, TGA a širokoúhlý rozptyl paprsků X (WAXS).
Exfoliovaná adsorpce je metoda přípravy nanokompozitů (NC) z
polymerního roztoku. Pro přípravu nanokompozitů byly využity PEA s
obsahem CLO jednotek 20-100%. Jako plnivo byly použity dva typy
organofilizovaného montmorillonitu (MMT) v koncentraci 5 hm% smektitu.
Byly získány folie o velikosti 8 x 12 cm a tloušťce cca 0,1 mm.
Úspěšnost přípravy PEA nanokompozitů metodou exfoliované adsorpce
závisí jak na použitém kopolymeru, tak na typu organického kationu v
MMT a polaritě rozpouštědla. Vhodnost zvolených rozpouštědel byla
testována botnáním modifikovaného MMT. Byl stanoven tzv. botnací index
a korelován se změnou vzdálenosti vrstev z WAXS měření. Tyto údaje
ukazují na schopnost rozpouštědla oddálit od sebe jednotlivé vrstvy
silikátu. Interakce polymer - rozpouštědlo byly studovány pomocí
viskozimetrických měření. PEA NC připravené exfoliovanou adsorpcí měly
většinou interkalovanou v některých případech exfoliovanou strukturu.
Další alternativou přípravy PEA nanokompozitů je polymerace in situ.
Je známo, že aniontovou polymerací CLA v přítomnosti organofilizovaného
MMT nelze v důsledku výměny alkylamoniových iontů za protionty
laktamátů připravit nanokompozity s exfoliovanou strukturou. Podobný
nežádoucí efekt lze předpokládat i při aniontové kopolymeraci CLA s
CLO. Proto jsme v naší práci využili alternativní způsob přípravy
PEA-NC. Inspirací nám byla úspěšná příprava PEA aniontovou polymerací
CLA v přítomnosti PCLO1 a příprava poly(ε-kaprolaktonových) nanokompozitů (PCLO-NC) s vysokým obsahem exfoliovaného plniva2. Postup přípravy PEA NC byl tedy rozdělen na dvě fáze: (i) na přípravu poly(ε-kaprolaktonových) nanokompozitů (PCLO-NC) s vysokým obsahem exfoliovaného plniva (ii)
následnou polymeraci CLA v přítomnosti připravených PCLO-NC. Tímto
způsobem připravené nanokompozity obsahovaly malý podíl neexfoliovaného
plniva v závislosti na experimentálních podmínkách přípravy a poměru
vstupních komponent.
Literatura:
- Bernášková A., Chromcová D., Brožek J., Roda J.: Polymer 45, 2141 (2004)
- Lepoittevin B., Pentoustier N., Decalckenaere M., Alexandre M., Kubies D., Calberg C., Jerome R., Dubois P.: Macromolecules 35, 8385 (2002)
Tato práce vznikla za podpory výzkumného záměru MSM 6046137302.