Main lectures
1 2 3
4 5 6 7 8 9 10
POLYMER-SOLVENT COMPLEXES AND INTERCALATES PREPARED UNDER
VARIOUS CONDITIONS
JEAN-MICHEL GUENET
Institut Charles Sadron CNRS UPR 22
6, rue Boussingault
F-67083 STRASBOURG Cedex FRANCE
guenet@ics.u-strasbg.fr
Polymer-solvent compounds (either complexes or intercalates)
can be prepared under various conditions: 1) by cooling
homogeneous polymer solutions, or 2) by exposing solid polymer
samples (films or bulk samples) to the liquid solvent or to its
vapours at a temperature well below the compound melting
temperature. One way of studying the outcomes of these different
ways of preparation is to establish the temperature-concentration
phase diagrams. After discussing the potentiality of these phase
diagrams for characterizing the systems under study, the results
will be presented on compounds involving isotactic and
syndiotactic polystyrenes. The various structures and
morphologies will be also presented. It will be particularly
shown that the path followed to reach a T,C coordinate of
the phase diagram (decreasing T at constant concentration C
or decreasing C at constant temperature T) has no
significant effect on the melting behaviour of the various
systems.
Microscopically-viewed Structural
Changes in Solvent-induced phase transitions of synthetic
polymers
K. TASHIRO
Graduate School of Science, Osaka
University, Toyonaka, Osaka 560-0043, Japan
Solvent-induced Crystallization of
syndiotactic Polystyrene
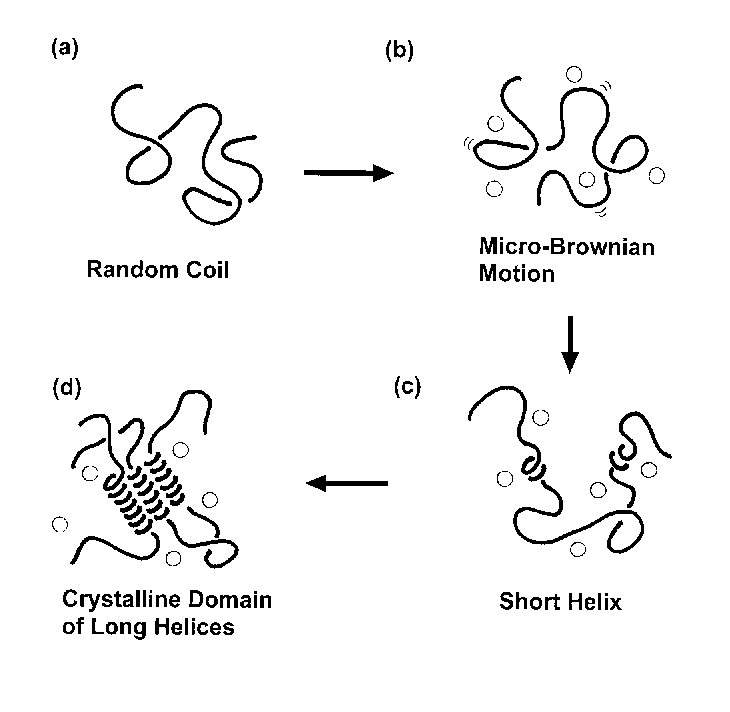 Syndiotactic polystyrene is known to crystallize
from the glassy state to the -form by absorbing organic solvent such as toluene,
benzene, chloroform, etc. This -form is a polymer-solvent complex, in which the
helical chains and solvent molecules are packed together. In
order to clarify the transition mechanism from the microscopic
point of view, we performed time-resolved measurements of X-ray
diffraction, infrared spectra and Raman spectra in this
solvent-induced crystallization process. On the basis of a
concept of critical sequence length for the infrared and Raman
bands, we could imagine concretely the growth of helical chains
and combined this conformational change with the formation of
crystal lattice as informed by X-ray diffraction data. The
quantitative analysis of half-width of infrared bands intrinsic
to the amorphous phase told us that the molecular motion of the
random coils is enhanced even below the glass transition
temperature through the interactions with solvent. By combining
all these information together, the structural formation process
could be clarified as shown in the above figure.
Syndiotactic polystyrene is known to crystallize
from the glassy state to the -form by absorbing organic solvent such as toluene,
benzene, chloroform, etc. This -form is a polymer-solvent complex, in which the
helical chains and solvent molecules are packed together. In
order to clarify the transition mechanism from the microscopic
point of view, we performed time-resolved measurements of X-ray
diffraction, infrared spectra and Raman spectra in this
solvent-induced crystallization process. On the basis of a
concept of critical sequence length for the infrared and Raman
bands, we could imagine concretely the growth of helical chains
and combined this conformational change with the formation of
crystal lattice as informed by X-ray diffraction data. The
quantitative analysis of half-width of infrared bands intrinsic
to the amorphous phase told us that the molecular motion of the
random coils is enhanced even below the glass transition
temperature through the interactions with solvent. By combining
all these information together, the structural formation process
could be clarified as shown in the above figure.
Water-induced Phase Transitions of
Poly(ethylene imine)
Poly(ethylene imine) shows a structural
change between a doubly-stranded helix in a dry state and a
planar-zigzag all-trans chain in humid state by supplying water
vapor. The zigzag chains form stoichiometric hydrates with water
through the intermolecular hydrogen bonds. Time-resolved
measurements of infrared spectra were carried out in the
hydration process and the detailed information could be obtained
as for the structural transformation between these anhydrate and
hydrates.
Highly conductive oriented Polymer electrolytes
D. Golodnitsky*, E. Livshits, E. Peled
School of Chemistry, Tel Aviv University, Tel Aviv 69978,
Israel
*E-mail:golod@post.tau.ac.il
The growing interest in the ion-conducting polymers is mainly
a result of the development of high quality processable and
environmentally stable materials for use in items such as
rechargeable batteries, electrochromic devices and sensors.
Moreover, polymer electrolytes are of theoretical interest as
model materials for the investigation of conduction paths in
semicrystalline systems.
It is generally acknowledged that the macroscopic properties
of polymeric materials are strongly influenced by their
structural organization, which in turn may be affected by
physical treatment. Molecular architecture plays an important
role in determining the ionic conductivity of polymer
electrolytes. It has been suggested that the ion transfer is
driven by competing inter- and intramolecular interactions. Ions
are thought to be transported by the quasi-random motion of short
polymer segments and the conductivity is generally observed to
rise with increasing flexibility of the polymer chains. In the
polymer-salt complexes of polyethylene oxide (PEO), cations are
enclosed within the PEO helices, thus suggesting the possibility
of efficient cation transport along the helical axis.
We have employed a variety of experimental methods, including
DC and AC conductivity, scanning electron microscopy (SEM),
atomic-force microscopy (AFM), X-ray diffraction, differential
scanning calorimetry (DSC), Fourier-transform infrared (FTIR)
spectroscopy, and pulsed field gradient nuclear magnetic
resonance (NMR) to investigate the effect of stretching in
poly(ethylene oxide) (PEO):LiX systems. Our studies show that
macroscopic stretch is fully reflected in the microscopic
distortion of the as-cast structure of polymer electrolytes. In
semicrystalline complexes of PEO with different salts, such as
lithium iodide, lithium trifluoromethanesulfonate, lithium
trifluoromethanesulfonimide, lithium hexafluoroarsenate and
lithium bis(oxalato)borate, enhancement of the stretching-induced
longitudinal DC conductivity by a factor of 5 to 40 was observed,
in spite of the formation of a more ordered polymer electrolyte
(PE) structure. It was found that the more amorphous the PE, the
less its lengthwise conductivity is influenced by stretching. The
results of our investigation support the idea of preferential
ionic transport along the PEO helical axis in the crystalline
phase and offer a fertile field for research and development in
the synthesis of new rigid polymers with ordered channels and
composition appropriate for enhanced ionic conductivity.
MOLECULAR TECTONICS: FROM MOLECULAR SIMPLICITY TO COMPLEX
MOLECULAR NETWORK
M. W. HOSSEINI
University Louis Pasteur, Institut Le Bel, 4, rue Blaise
Pascal, 67000 Strasbourg, France, hosseini@chimie.u-strasbg.fr
The development of concepts and strategies allowing the design
of molecular networks with predicted and programmed structures is
a topic of active investigations in research groups throughout
the world. The engineering of molecular networks involves
modeling, synthesis and exploitation of crystalline solids with
predefined connectivity patterns between molecules or ions. As
such, molecular network engineering, a subset of crystal
engineering, is at the intersection of supramolecular chemistry
and materials chemistry.
What has been accomplished: Structural molecular networks:
Molecular networks are defined as supramolecular structures
possessing translational symmetry. Whereas molecules are
described as assemblies of atoms interconnected by covalent
bonds, by analogy, one may describe molecular networks as
hypermolecules for which the connectivity between the elementary
molecular components (tectons) is ensured by non-covalent and/or
reversible interactions. The formation of these molecular solids
results from the chemical and physical nature of the molecular
components comprising the solid. The design and formation of
large molecular networks (10-6-10-3 m scale), may hardly be
envisaged through a step-by-step type strategy. However, the
preparation of such higher-order materials may be achieved
through iterative self-assembling processes that engage
programmed tectons. Thus, by programming (storage of both
recognition and iteration information) specific interaction
patterns between tectons in the crystalline phase, a variety of
molecular (hydrogen bonded-, inclusion-, coordination-) networks
with various dimensionality may be designed.
What remains to be accomplished: Functional molecular
networks: Thus far, the approaches employed are mainly
concerned with structural control of the formation of molecular
networks. Obviously, a further step, which remains to be
achieved, will be to use the structural knowledge gathered over
the last decade for the design of functional networks. One may
foresee that the majority of molecular materials for the next
century might be based on functional molecular networks. In
particular, using physical properties such magnetism,
conductivity, optical and/or electronic features, new materials
with applications in the field of storage, sensing, signal
treatment, transduction and decontamination will be designed and
prepared.
ON THE STRUCTURE OF SULFONIC ACID DOPED
THERMOREVERSIBLE POLYANILINE GELS
TUSHAR JANA, JHUNU CHATTERJEE, ARUN K. NANDI*
Polymer Science Unit, Indian Association For The Cultivation
Of Science Jadavpur, Calcutta - 700032, INDIA, E-mail:
psuakn@mahendra.iacs.res.in
The surface morphology of the thermoreversible polyaniline
(PANI) gels prepared with dinonylnaphthalene sulfonic acid
(DNNSA), dinonylnaphthalene disulfonic acid (DNNDSA), (±)
camphor-10 - sulfonic acid (CSA) and n-dodecyloxo sulfonic acid
(DOSA) are studied using atomic force microscopy (AFM) for 15%
PANI concentration (w/w). The AFM study clearly reveals the
formation of lamellar morphology in the gel. A representative AFM
picture showing lamellar morphology is presented in Fig.-1 for
the PANI - DNNSA system. X -ray scattering experiments of these
gels also support the lamellar structure formation and the
lamellar thickness measured from it remains invariant with
composition for the PANI - DNNSA and PANI - DNNDSA systems. In
PANI - CSA and in PANI -DOSA systems the lamellar thicknesses
vary with PANI concentration. Both X ray diffraction and electron
diffraction experiments on the gels of different compositions of
PANI - DNNSA and PANI - DNNDSA systems indicate new spacings of
lower dhkl values which are invariant with
composition. Similar observations are also found in PANI - CSA
and in PANI - DOSA systems. These results indicate the formation
of new unit cells in the lamella due to the microcrystallization
of elongated surfactant tails formed under the doped condition.
The dhkl values characterizing the lamellar thickness,
bilayer thickness and monolayer thickness are discussed in view
of molecular mechanics calculations using MMX program. The
composition dependency of the lamellar thickness is discussed
from the cohesive force of the surfactant tails within the
lamella.

Fig. 1
CONFORMATIONAL CHARACTERISTICS OF STEREOREGULAR
POLY(METHYL METHACRYLATE)S AND OF THE STEREOCOMPLEX: NEW INSIGHTS
INTO THE BULK AND SURFACE STRUCTURE FROM FTIR MEASUREMENTS
O. N. TRETINNIKOV
B. I. Stepanov Institute of Physics, National Academy of
Sciences of Belarus, Prospekt F. Skariny 70, Minsk, 220072,
Belarus, olegtret@dragon.bas-net.by
Stereoregular PMMA (isotactic (i-PMMA) and syndiotactic
(s-PMMA)) polymers display the strongest effect of tacticity on
physical properties among all strereoregular polymers studied so
far. I-PMMA and s-PMMA differ markedly in the glass transition
temperature, chain stiffness, miscibility, surface activity, and
adsorption behavior. The fact that these physically unlike
macromolecules strongly attract each other with the resultant
formation of stereocomplexes is really remarkable. Although
stereoregular PMMAs and the stereocomplex have been the subj ect
of intensive studies over the last three decades, their
conformational characteristics are still a matter of debate and
controversy.
Herein, we contribute to this field by variable-temperature
FTIR measurements on amorphous films of isotactic and
syndiotactic PMMA, followed by a detailed analysis of the
temperature dependence of integrated band intensities for C-O
stretching modes in the region 1050-1300 cm-1. The use
of computerized FTIR instrumentation and modern methods of
quantitative band-fitting analysis in combination with the recent
IR and ab initio results for simple esters allowed us to achieve,
for the first time, the unambiguous conformational assignment for
the C-O bands of PMMA. This, in turn, enabled reliable IR
spectroscopic determination of the difference in the energy of
accessible conformational states for the backbone and for the
ester group. Finally, detailed analyses of the C-O band
intensities in transmission FTIR and ATR-FTIR spectra of
single-component and stereocomplex PMMA films, based on the
established band assignments, revealed new features in the bulk
and surface structure of these important materials.
In printed form only
REORGANIZATION OF THE STRUCTURES, MORPHOLOGIES, AND CONFORMATIONS
OF POLYMERS BY COALESCENCE FROM THEIR CRYSTALLINE INCLUSION COMPOUNDS
FORMED WITH CYCLODEXTRINS
ALAN E. TONELLI*
Fiber & Polymer Science Program, North Carolina State
University, Raleigh, NC 27695-8301, USA alan_tonelli@ncsu.edu
We and several other research groups have recently reported
the ability of cyclodextrins (CDs) to act as hosts in the
formation of inclusion compounds (ICs) with guest polymers.
Polymer-CD-ICs are crystalline materials formed by the close
packing of host CD stacks, which results in a continuous channel
of ~5-10 A. in diameter running down the interior of the CD
stacks. The guest polymers are confined to the narrow, continuous
CD channels, and so are necessarily highly extended and
segregated from neighboring polymer chains by the walls of the CD
stacks. We have shown that coalescence of guest polymers from
their CD-IC crystals can result in a significant reorganization
of the structures, morphologies, and even conformations that are
normally observed in their bulk samples. For example, when
poly(ethylene terephthalate) (PET) is coalesced from its g-CD-IC, we find that in the non-crystalline
regions of the sample the PET chains are adopting highly extended
kink conformations, which result in their facile
recrystallization from the melt and prevent quenching of the
coalesced PET to achieve an amorphous sample during rapid cooling
from above Tm. We have also created well-mixed blends
of normally incompatible polymers by coalescing them from CD-ICs
containing both polymers, where they are necessarily spatially
proximal. Finally we have found the unique morphologies created
by the coalescence of homopolymers, block copolymers, and
homopolymer pairs from their CD-ICs are generally stable ta heat
treatment for prolonged periods above their Tm's and/or
Tg's, and so may be thermoplastically processed
without loss of the unique morphologies achieved through
coalescence from their CD-I~ crystals.
* The following colleagues (C. C. Rusa, X. Shuai, T. A. Bullions,
C. M. Balik, D. I Shin, and J.L. White) and students (M. Wei, F.
E. Porbeni, M. J. Gerber, E.Edeki, W. davis, and B. Urban) have
been active collaborators and the U.S. Dept. of Commerce (National
Textile Center) and U.S. Army (ARO) have generousty sponsored this
research.
WATER-PROTEIN AND LIGAND-PROTEIN INTERACTIONS
AS DETERMINED BY SELECTIVE NMR RELAXATION STUDIES
C. ROSSI*, S. MARTINI, M. RICCI, M.P. PICCHI AND C. BONECHI
Department of Chemical and Biosystem Sciences,
University of Siena, Via Aldo Moro,2 - 53100 Siena, Italy
- e-mail: rossi@unisi.it
Water-macromolecule and ligand-macromolecule interactions are
different phenomena and have different chemical and biological
importance. However both solvent-macromolecule and
ligand-macromolecule interaction can be studied using similar
approaches based on selective NMR relaxation experiments.
Macromolecules in solution induce solvent-solute interactions.
The extent of interaction can be studied by checking the solvent
parameters mostly affected by the presence of a large, slowly
reorienting biomacromolecule. Water proton relaxation rates have
been used to investigate different systems and phenomena, and
theoretical interpretations of the experimental results have been
proposed. In this paper both the water proton spin-lattice
relaxation rates  and
and  are analyzed
considering all possible sources of dipolar contributions. On the
basis of the discussed equations, the average effective
rotational water correlation time was calculated. The c value obtained in the binary
water-macromolecule system is then compared with the value
obtained in the water-macromolecule-ligand ternary system. The
result shows that this approach could be used as a parameter to
monitor the extent of solute-solvent interaction and to detect
the effects of the ligand molecules on receptor proteins as a
consequence of the recognition process. Selective and non-selective spin-lattice
relaxation rates were also used to investigate
ligand-macromolecules interactions. Several experimental and
theoretical approaches have been developed to study the
recognition processes between ligands and receptors, including
methods based on Nuclear Magnetic Resonance (NMR) analysis of the
solution behaviour of the ligand in the presence of receptors. In
this case the analysis is based on the comparison of selective (
are analyzed
considering all possible sources of dipolar contributions. On the
basis of the discussed equations, the average effective
rotational water correlation time was calculated. The c value obtained in the binary
water-macromolecule system is then compared with the value
obtained in the water-macromolecule-ligand ternary system. The
result shows that this approach could be used as a parameter to
monitor the extent of solute-solvent interaction and to detect
the effects of the ligand molecules on receptor proteins as a
consequence of the recognition process. Selective and non-selective spin-lattice
relaxation rates were also used to investigate
ligand-macromolecules interactions. Several experimental and
theoretical approaches have been developed to study the
recognition processes between ligands and receptors, including
methods based on Nuclear Magnetic Resonance (NMR) analysis of the
solution behaviour of the ligand in the presence of receptors. In
this case the analysis is based on the comparison of selective ( ) and non-selective (
) and non-selective ( ) spin-lattice
relaxation rate analysis of the ligand in the presence and
absence of macromolecular receptors and on the
) spin-lattice
relaxation rate analysis of the ligand in the presence and
absence of macromolecular receptors and on the  and
and  temperature dependence
analysis. From these studies, the ligand-receptor binding
strength was defined on the basis of the ”affinity index”
temperature dependence
analysis. From these studies, the ligand-receptor binding
strength was defined on the basis of the ”affinity index” . An index derived on
the basis of specific consideration about the
macromolecule-ligand equilibrium kinetics.
. An index derived on
the basis of specific consideration about the
macromolecule-ligand equilibrium kinetics.
Force spectroscopy studies of complexes with single polymer
molecules
M.J. Miles
H.H. Wills Physics Laboratory, University of Bristol, Tyndall
Avenue, Bristol BS8 1TL, United Kingdom
Atomic force microscopy (AFM) has been relatively little used
in the study of polymer-solvent interactions. However, it has
great potential to provide complementary information to existing
techniques in this field. In this presentation, four AFM
techniques which appear to be most promising will be presented
and some examples will be given.
The areas in which AFM measurements can contribute to the
study of polymer-solvent complexes are: (i) imaging, (ii) force
recognition, (iii) force spectroscopy, and (iv) microcantilever
sensors.
- AFM is capable of imaging single molecules at high
resolution. From such images, it is possible to determine
the flexibility and persistence length of individual
molecules in the environments of various solvents.
High-resolution imaging might be capable of following
structural changes in, for example, the helical repeat or
molecular width as a function of the solvent environment.
It might even been possible to image individual complexed
solvent molecules, but this would certainly be extremely
demanding technically. The related technique of scanning
near-field optical microscopy might also make a
contribution by providing optical information beyond the
diffraction limit.
- Force recognition involves functionalizing the AFM tip
with a test molecule, for example, a solvent molecule,
and testing its interaction against a polymer molecule.
In this way, the interaction forces, and ultimately the
binding energies of such interactions might be measured.
- Force spectroscopy is a single molecule technique in
which a polymer molecule is extended between a fixed
surface and the AFM tip. A force-extension curve is
obtained. Such measurements could be performed with and
without the presence of a particular solvent and the
change in mechanical properties of complexation
determined. During the extension, changes in the
helical/secondary structure can be detected and the used
of the dynamic version of this technique allows the
complex mechanical properties to be measured during
transitions. This is the most promising technique and
some data on the effect of complexation will be
presented.
- AFM technology is proving to be very valuable in the
development of new micro sensors. In this technique, an
AFM force-sensing cantilever is coated with polymer
molecules and the response of the cantilever to the
presence of solvent molecules can provide information on
the nature of the interaction.
FUNCTIONAL POLYMERIC MATERIALS: COMPLEXING ADDITIVES AS
STRUCTURE-INDUCING ELEMENTS
G. TEN BRINKEa, O. IKKALAb
aLaboratory of Polymer Chemistry, Materials Science
Centre, University of Groningen, Nijenborgh 4, 9747 AG Groningen,
The Netherlands
bDepartment of Engineering Physics and Mathematics,
and Center for New Materials, Helsinki University of Technology,
P.O. Box 2200, FIN-02015 HUT, Finland
Self-assembly of polymeric supramolecules obtained by
recognition driven supramolecule formation in polymers is a very
powerful tool to produce functional polymer materials. The
concept is illustrated using comb-shaped architectures. The
physically bonded side chains can have two separate ”functions”.
For example, in addition to providing a repulsive side chain
required for self-organization, they may contain an acidic group
that simultaneously acts as a dopant for a conjugated polymer
such as polyaniline, which leads to electronic conductivity.
Another example involves polarized luminance in solid state films
of rod-like polymers obtained by removing the hydrogen bonded
side chains from the thermotropic smectic phase. To increase
complexity, one can incorporate structural hierarchies. This can
be accomplished by applying within a single material different
self-organization and recognition mechanisms operating at
different length scales. For example, block copolymeric
self-organization at the 100-2000 Å length scale and
polymer/amphiphiles self-organization at the 10-60 Å length
scale can be combined. Upon selective ”doping” of one block,
conductivity can be switched based on a sequence of phase
transition. Macroscopically tridirectional protonic conductivity
can be accomplished by flow-orienting the local structures. There
is a rich variety of phases, eg. lamellae-within-cylinders,
which also allow selective cleaving of the constituents which
form the supramolecules. For example, starting from polystyrene-block-poly(4-vinyl
pyridine) where pentadecylphenol has been hydrogen bonded, one
obtains a structure where the glassy polystyrene matrix contains
empty cylindrical pores with poly(4-vinylpyridine) brushes at the
walls. By selecting different block copolymers and amphiphiles,
one can tune the wettability of the pore walls. In principle even
the conformations of the brushes can be controlled by selecting
the polymer and solvent properly. Finally, selective cleaving of
the constituents, which form the supramolecules, also allows the
preparation of a variety of nano-objects.