
Polymer-Antimicrobial Peptide Constructs with Tailored Drug-Release Behavior
 , , , , ,
, , , , ,
Abstract
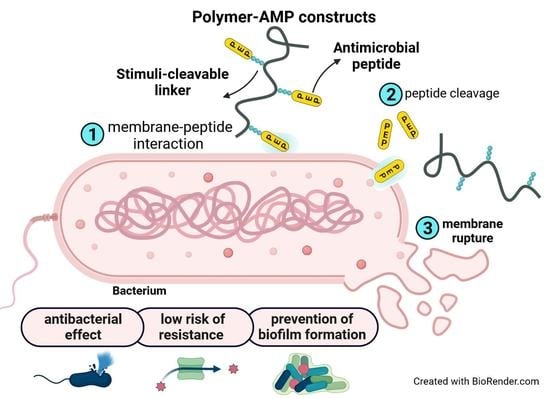
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
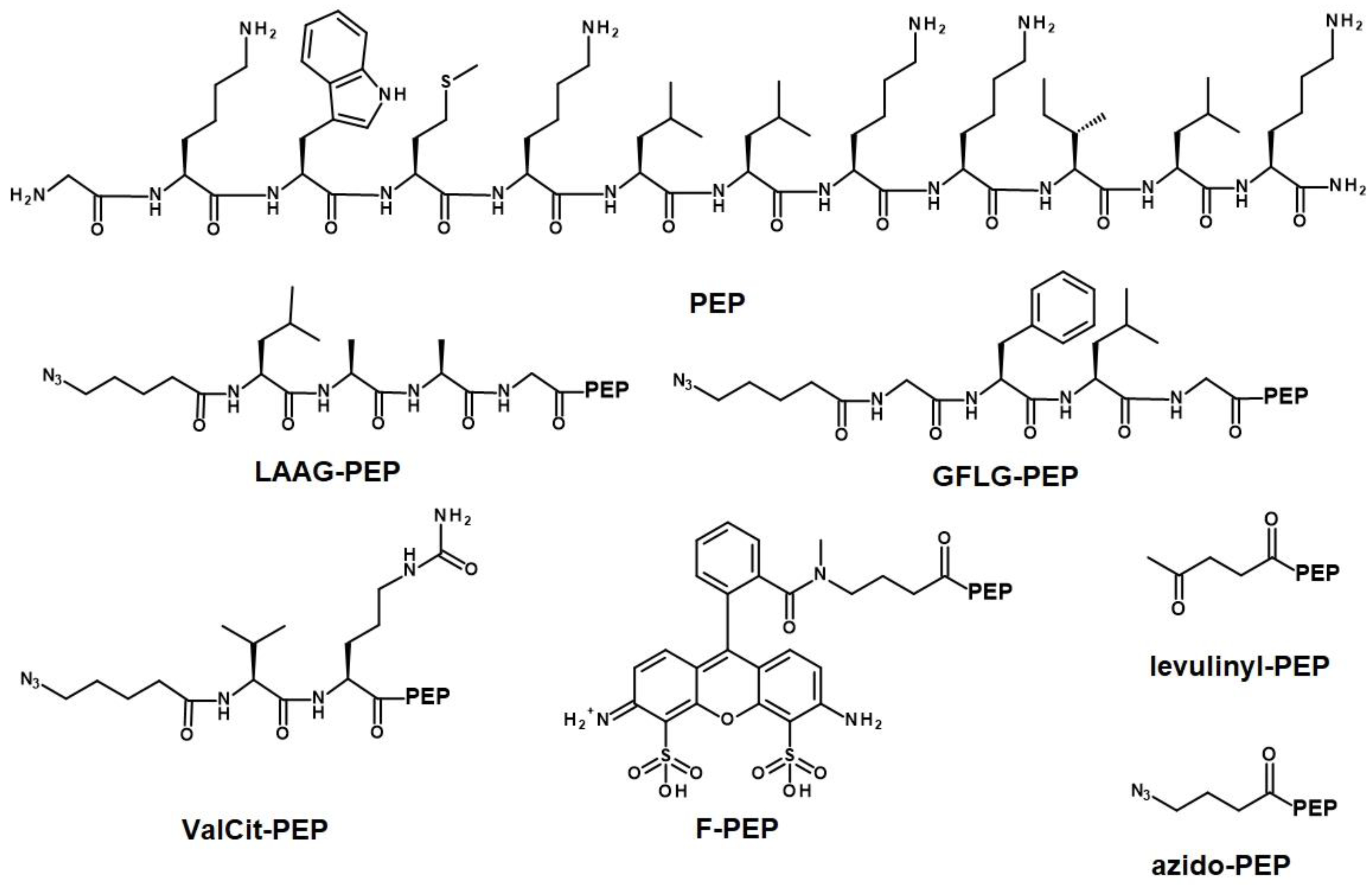
2.3. Synthesis of Peptide Derivatives
2.4. Synthesis of the Monomers
2.5. Synthesis of the Polymer Precursor
2.6. Synthesis of the Polymer Conjugates with Azido-Peptides
2.7. Synthesis of the Polymer P-Hyd-PEP with Keto Group Containing Levulinyl-PEP
2.8. Stability and Release of the PEP
2.9. Determination of Minimal Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC)
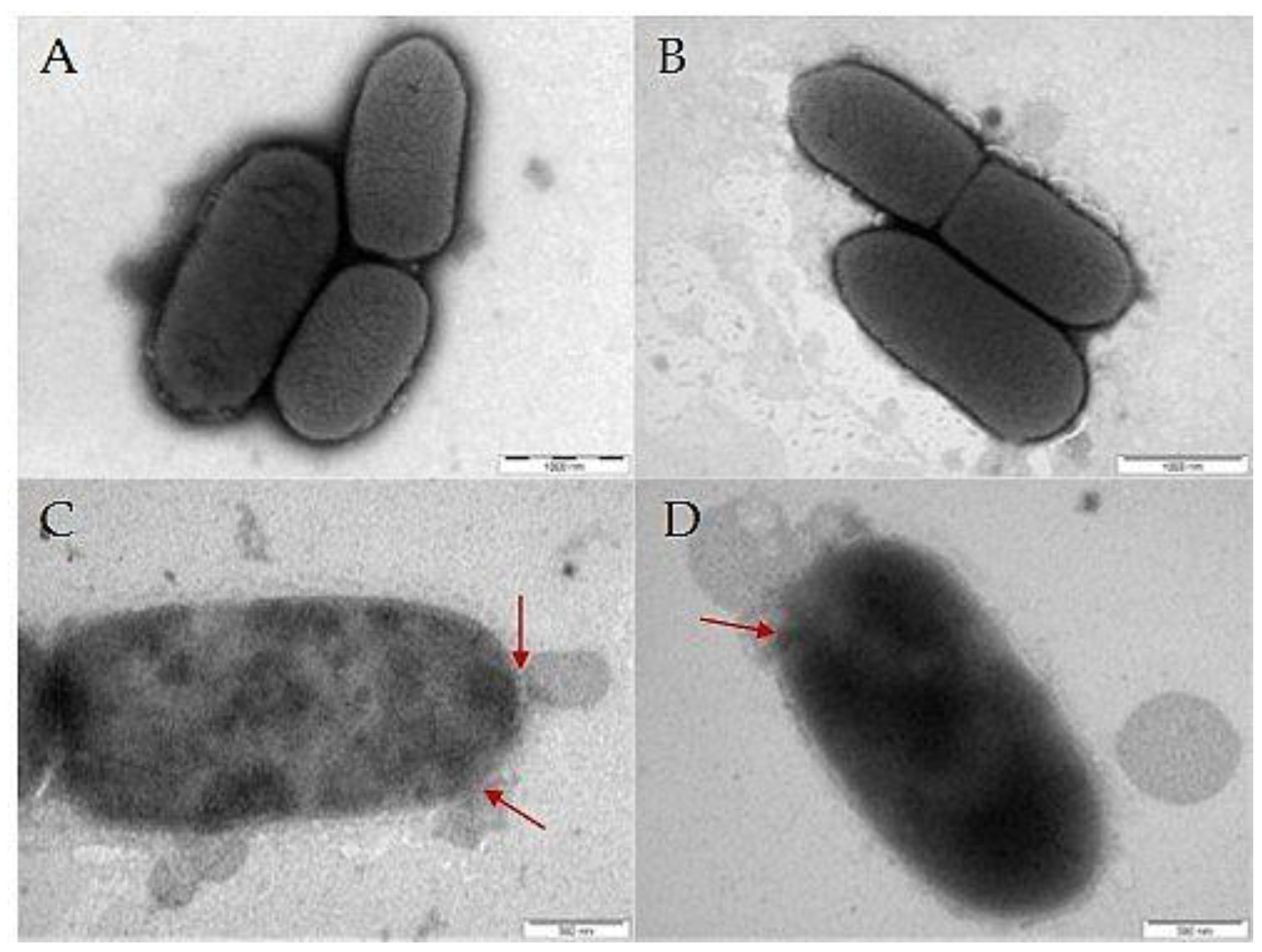
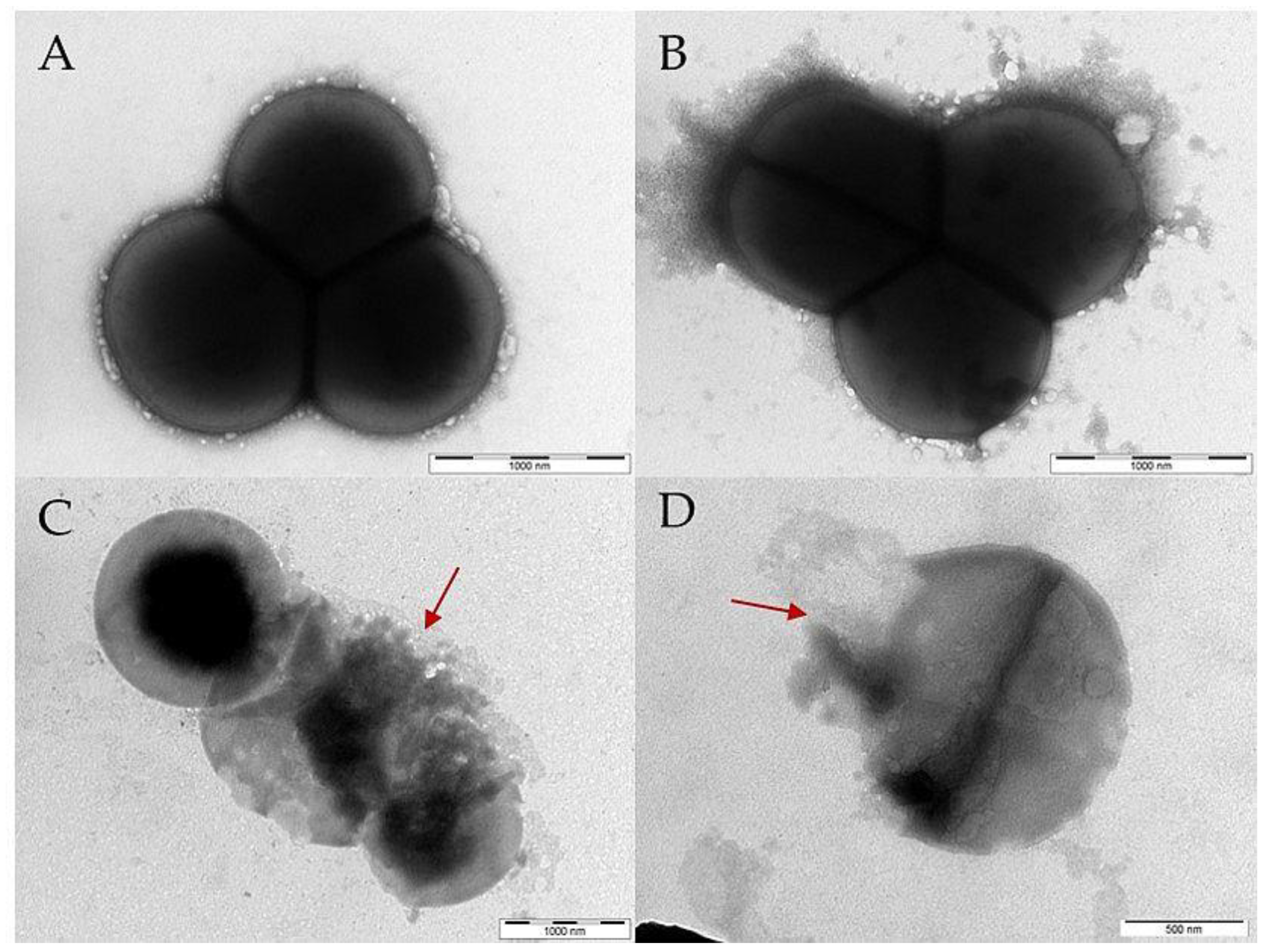
2.10. Transmission Electron Microscopy (TEM)
2.11. Fluorescent Microscopy
3. Results and Discussion
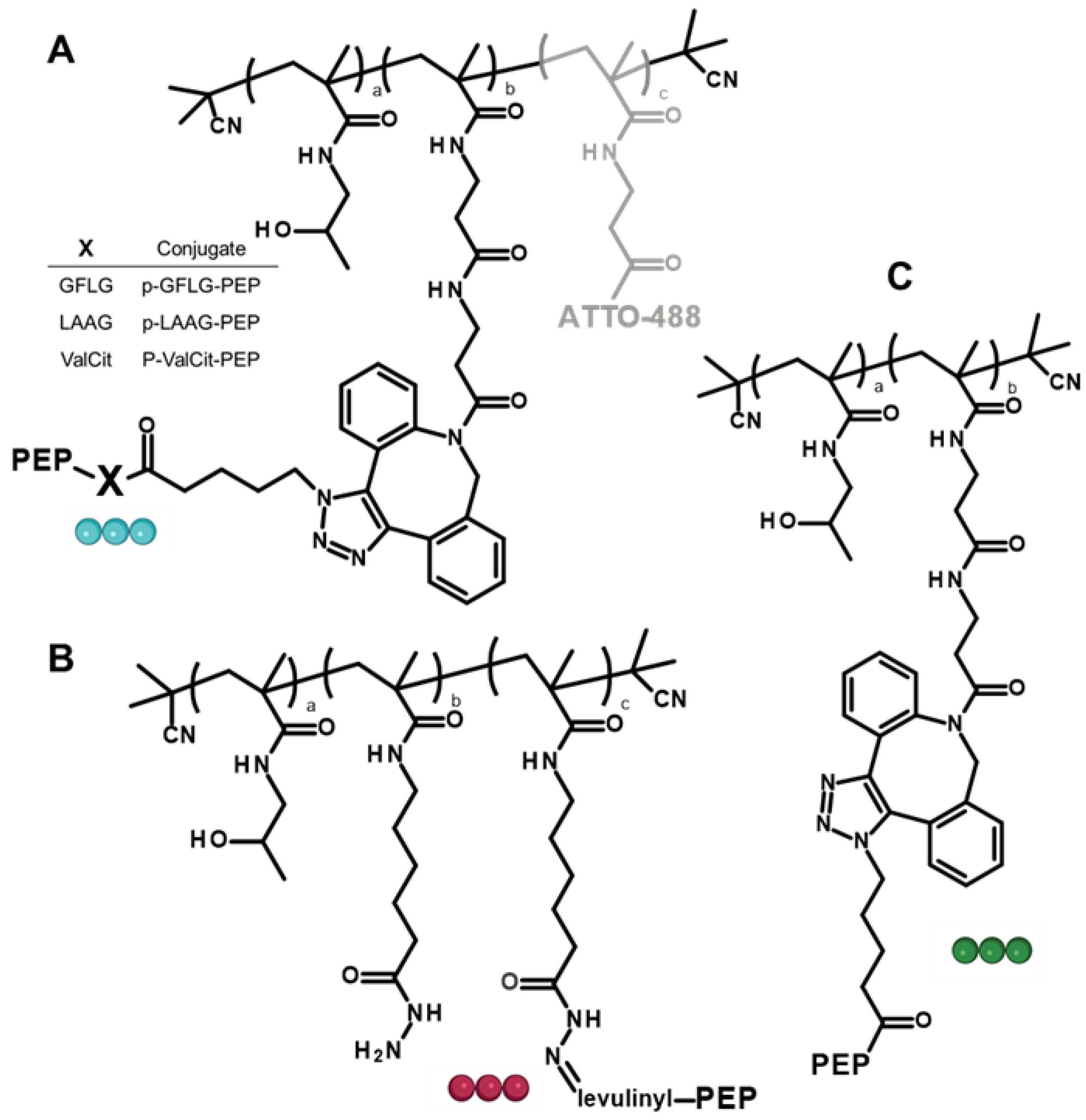
3.1. Synthesis of Polymer Precursors
3.2. Synthesis of Polymer-Peptide Constructs
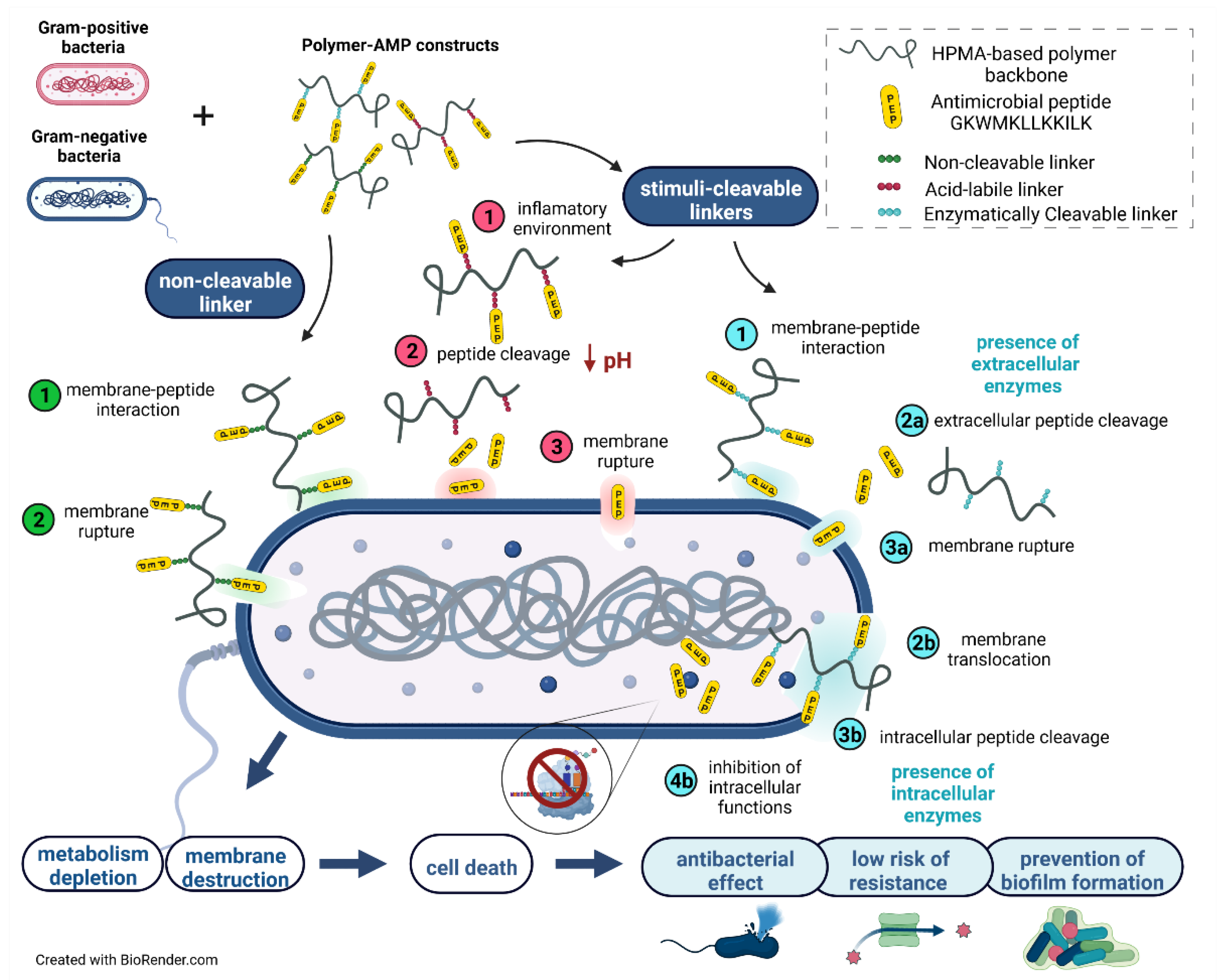
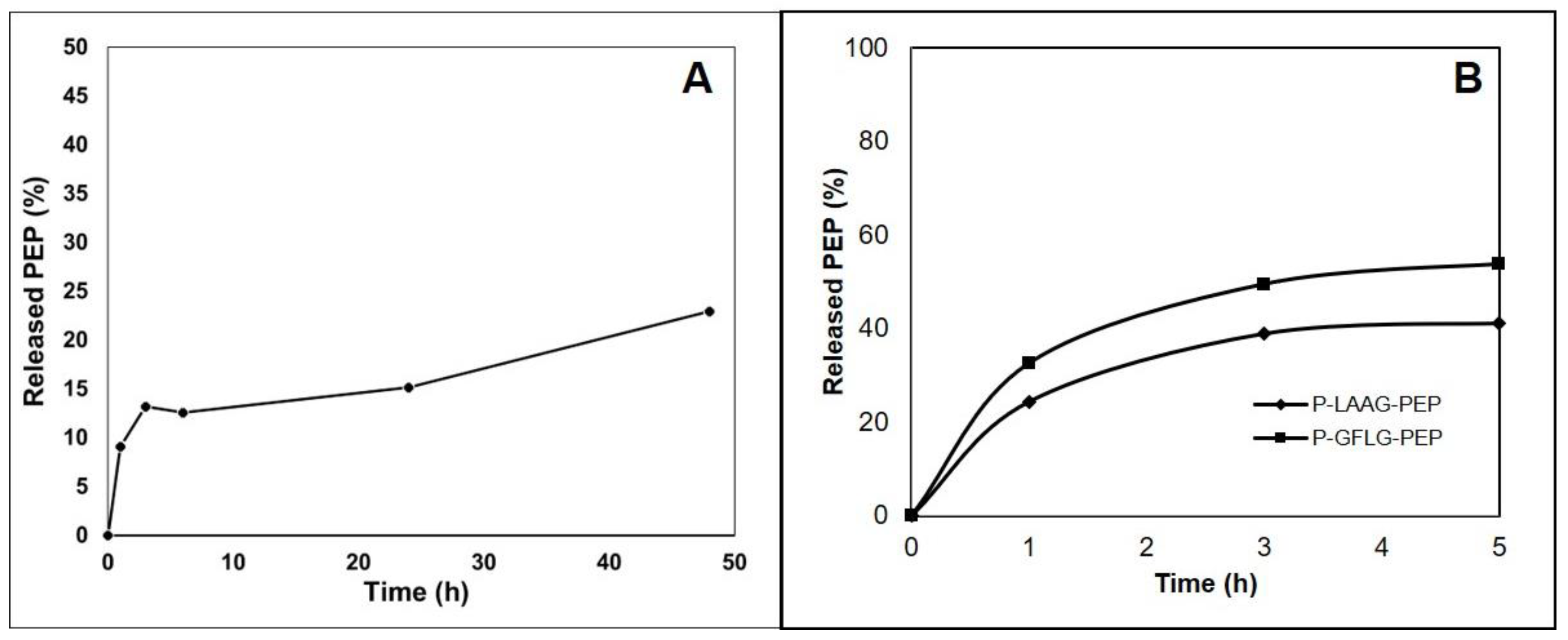
3.3. Design of the Polymer-AMP Constructs, AMP Release, and Stability
3.4. Minimum Inhibitory Concentration (MIC) of PEP and PEP-Polymer Constructs
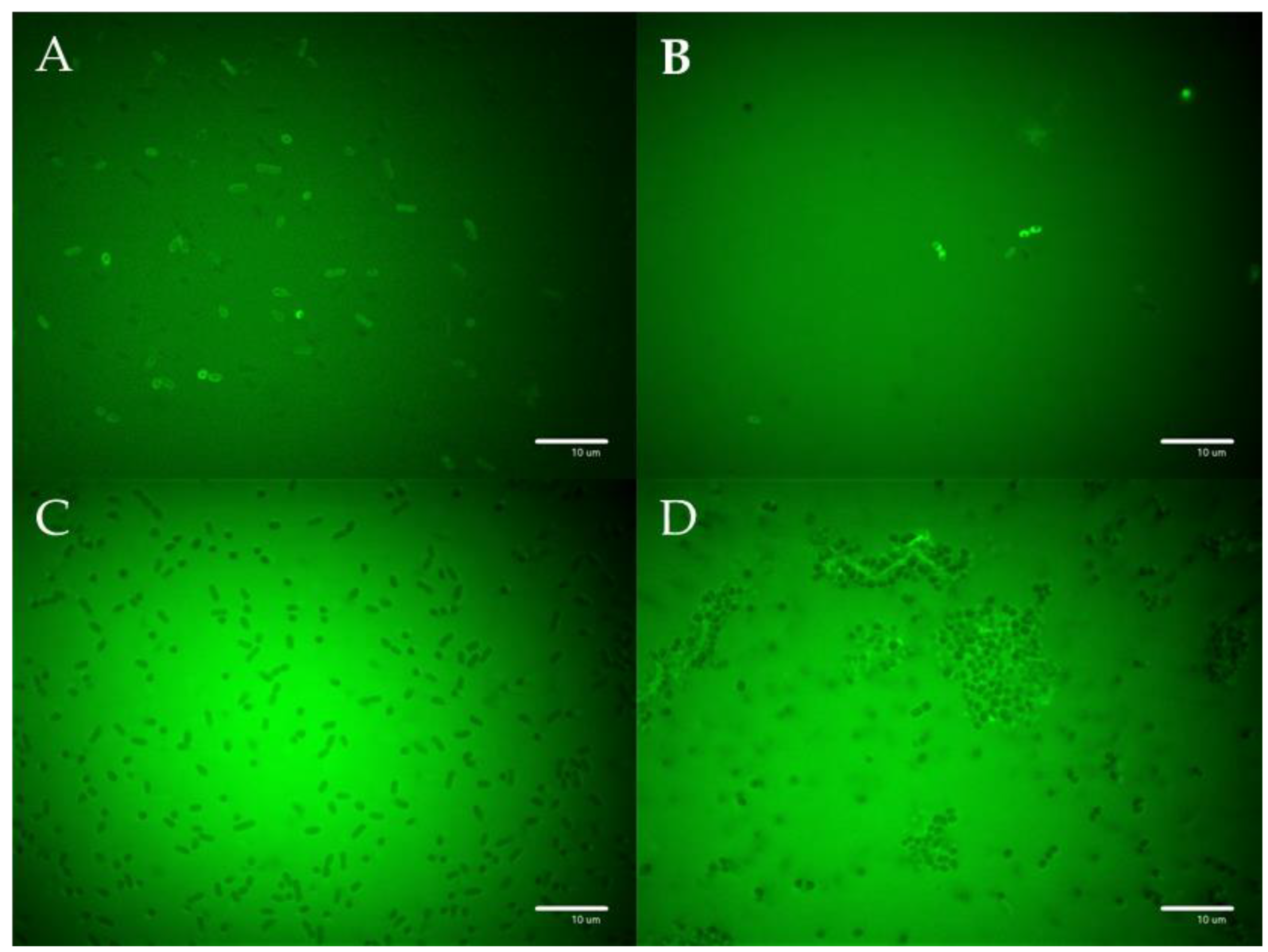
3.5. Visualization of the PEP and PEP Construct’s Effect on Selected Bacterial Strains
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Seaworth, B.J.; Griffith, D.E. Therapy of Multidrug-Resistant and Extensively Drug-Resistant Tuberculosis. Microbiol. Spectr. 2017, 5, 1–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Global Tuberculosis Report 2021; World Health Organization: Geneva, Switzerland, 2021; ISBN 9789240037021.
- Mintz, P.D. WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed Thereby Highlighting the Requirement for Further Vigilance for Platelet Transfusions; World Health Organization: Geneva, Switzerland, 2017; pp. 3–4.
- Asokan, G.; Ramadhan, T.; Ahmed, E.; Sanad, H. WHO Global Priority Pathogens List: A Bibliometric Analysis of Medline-PubMed for Knowledge Mobilization to Infection Prevention and Control Practices in Bahrain. Oman Med. J. 2019, 34, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Griffith, D.E.; Aksamit, T.R. Understanding nontuberculous mycobacterial lung disease: It’s been a long time coming. F1000Research 2016, 5, 2797–2805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cândido, P.H.C.; De Nunes, L.S.; Marques, E.A.; Folescu, T.W.; Coelho, F.S.; De Moura, V.C.N.; Da Silva, M.G.; Gomes, K.M.; Lourenço, M.C.D.S.; Aguiar, F.S.; et al. Multidrug-resistant nontuberculous mycobacteria isolated from cystic fibrosis patients. J. Clin. Microbiol. 2014, 52, 2990–2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zasloff, M. Antimicrobial peptides of multicellularorganisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Bulet, P.; Stöcklin, R.; Menin, L. Anti-microbial peptides: From invertebrates to vertebrates. Immunol. Rev. 2004, 198, 169–184. [Google Scholar] [CrossRef]
- Hale, J.D.F.; Hancock, R.E.W. Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev. Anti. Infect. Ther. 2007, 5, 951–959. [Google Scholar] [CrossRef]
- Steckbeck, J.D.; Deslouches, B.; Montelaro, R.C. Antimicrobial peptides: New drugs for bad bugs? Expert Opin. Biol. Ther. 2014, 14, 11–14. [Google Scholar] [CrossRef] [Green Version]
- Vicent, M.J.; Manzanaro, S.; de la Fuente, J.A.; Duncan, R. HPMA copolymer-1,5-diazaanthraquinone conjugates as novel anticancer therapeutics. J. Drug Target. 2004, 12, 503–515. [Google Scholar] [CrossRef]
- Fox, J.L. Antimicrobial peptides stage a comeback. Nat. Biotechnol. 2013, 31, 379–382. [Google Scholar] [CrossRef]
- Monincová, L.; Buděšínsky, M.; Slaninová, J.; Oldřich Hovorka, O.; Cvačka, J.; Voburka, Z.; Fučík, V.; Borovičková, L.; Bednárová, L.; Straka, J.; et al. Novel antimicrobial peptides from the venom of the eusocial bee Halictus sexcinctus (Hymenoptera: Halictidae) and their analogs. Amino Acids 2010, 39, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Melicherčík, P.; Nešuta, O.; Čeřovský, V. Antimicrobial Peptides for Topical Treatment of Osteomyelitis and Implant-Related Infections: Study in the Spongy Bone. Pharmaceuticals 2018, 11, 20. [Google Scholar] [CrossRef] [Green Version]
- Ulbrich, K.; Holá, K.; Šubr, V.; Bakandritsos, A.; Tuček, J.; Zbořil, R. Targeted Drug Delivery with Polymers and Magnetic Nanoparticles: Covalent and Noncovalent Approaches, Release Control, and Clinical Studies. Chem. Rev. 2016, 116, 5338–5431. [Google Scholar] [CrossRef] [Green Version]
- Letai, A. S63845, an MCL-1 Selective BH3 Mimetic: Another Arrow in Our Quiver. Cancer Cell 2016, 30, 834–835. [Google Scholar] [CrossRef] [Green Version]
- Subr, V.; Etrych, T.; Ulbrich, K.; Hirano, T.; Kondo, T.; Todoroki, T.; Jelinkova, M.; Rihova, B. Synthesis and properties of poly{[}N-(2-hydroxypropyl) methacrylamide] conjugates of superoxide dismutase. J. Bioact. Compat. Polym. 2002, 17, 105–122. [Google Scholar]
- Tang, Z.; Ma, Q.; Chen, X.; Chen, T.; Ying, Y.; Xi, X.; Wang, L.; Ma, C.; Shaw, C.; Zhou, M. Recent Advances and Challenges in Nanodelivery Systems for Antimicrobial Peptides (AMPs). Antibiotics 2021, 10, 990. [Google Scholar] [CrossRef] [PubMed]
- Cayot, P.; Tainturier, G. The quantification of protein amino groups by the trinitrobenzenesulfonic acid method: A reexamination. Anal. Biochem. 1997, 249, 184–200. [Google Scholar] [CrossRef]
- Ulbrich, K.; Šubr, V.; Strohalm, J.; Plocová, D.; Jelínková, M.; Říhová, B. Polymeric drugs based on conjugates of synthetic and natural macromolecules. I. Synthesis and physico-chemical characterisation. J. Control. Release 2000, 64, 63–79. [Google Scholar] [CrossRef]
- Pola, R.; Parnica, J.; Zuska, K.; Böhmová, E.; Filipová, M.; Pechar, M.; Pankrác, J.; Mucksová, J.; Kalina, J.; Trefil, P.; et al. Oligopeptide-targeted polymer nanoprobes for fluorescence-guided endoscopic surgery. Multifunct. Mater. 2019, 2, 024004. [Google Scholar] [CrossRef]
- Chytil, P.; Etrych, T.; Kříž, J.; Subr, V.; Ulbrich, K. N-(2-Hydroxypropyl)methacrylamide-based polymer conjugates with pH-controlled activation of doxorubicin for cell-specific or passive tumour targeting. Synthesis by RAFT polymerisation and physicochemical characterisation. Eur. J. Pharm. Sci. 2010, 41, 473–482. [Google Scholar] [CrossRef]
- Pola, R.; Janoušková, O.; Etrych, T. The pH-Dependent and Enzymatic Release of Cytarabine from Hydrophilic Polymer Conjugates. Physiol. Res. 2016, 65, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Perrier, S.; Takolpuckdee, P.; Westwood, J.; Lewis, D.M. Versatile Chain Transfer Agents for Reversible Addition Fragmentation Chain Transfer (RAFT) Polymerization to Synthesize Functional Polymeric Architectures. Macromolecules 2004, 37, 2709–2717. [Google Scholar] [CrossRef]
- Chytil, P.; Koziolová, E.; Etrych, T.; Ulbrich, K. HPMA Copolymer–Drug Conjugates with Controlled Tumor-Specific Drug Release. Macromol. Biosci. 2018, 18, 1700209. [Google Scholar] [CrossRef] [PubMed]
- Kopeček, J.; Kopečková, P. HPMA copolymers: Origins, early developments, present, and future. Adv. Drug Deliv. Rev. 2010, 62, 122–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randárová, E.; Kudláčová, J.; Etrych, T. HPMA copolymer-antibody constructs in neoplastic treatment: An overview of therapeutics, targeted diagnostics, and drug-free systems. J. Control. Release 2020, 325, 304–322. [Google Scholar] [CrossRef]
- Zdzalik, M.; Karim, A.Y.; Wolski, K.; Buda, P.; Wojcik, K.; Brueggemann, S.; Wojciechowski, P.; Eick, S.; Calander, A.-M.; Jonsson, I.-M.; et al. Prevalence of genes encoding extracellular proteases in Staphylococcus aureus—Important targets triggering immune response in vivo. FEMS Immunol. Med. Microbiol. 2012, 66, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalińska, M.; Kantyka, T.; Greenbaum, D.C.; Larsen, K.S.; Władyka, B.; Jabaiah, A.; Bogyo, M.; Daugherty, P.S.; Wysocka, M.; Jaros, M.; et al. Substrate specificity of Staphylococcus aureus cysteine proteases—Staphopains A, B and C. Biochimie 2012, 94, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Pechar, M.; Pola, R.; Studenovský, M.; Bláhová, M.; Grosmanová, E.; Dydowiczová, A.; Filipová, M.; Islam, R.; Gao, S.; Fang, J.; et al. Polymer nanomedicines with enzymatically triggered activation: A comparative study of in vitro and in vivo anti-cancer efficacy related to the spacer structure. Nanomed. Nanotechnol. Biol. Med. 2022, 46, 9–12. [Google Scholar] [CrossRef]
- Volejníková, A.; Melicherčík, P.; Nešuta, O.; Vaňková, E.; Bednárová, L.; Rybáček, J.; Čeřovský, V. Antimicrobial peptides prevent bacterial biofilm formation on the surface of polymethylmethacrylate bone cement. J. Med. Microbiol. 2019, 68, 961–972. [Google Scholar] [CrossRef]
- Sánchez-Clemente, R.; Igeño, M.I.; Población, A.G.; Guijo, M.I.; Merchán, F.; Blasco, R. Study of pH Changes in Media during Bacterial Growth of Several Environmental Strains. Proceedings 2018, 2, 1297. [Google Scholar]
- Wilhelm, M.J.; Sharifian Gh., M.; Wu, T.; Li, Y.; Chang, C.-M.; Ma, J.; Dai, H.-L. Determination of bacterial surface charge density via saturation of adsorbed ions. Biophys. J. 2021, 120, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Šálek, P.; Trousil, J.; Nováčková, J.; Hromádková, J.; Mahun, A.; Kobera, L. Poly [2-(dimethylamino)ethyl methacrylate- co -ethylene dimethacrylate]nanogel by dispersion polymerization for inhibition of pathogenic bacteria. RSC Adv. 2021, 11, 33461–33470. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Structure | Content of Reactive Group mol% | Mw g‧mol−1 | Ð |
|---|---|---|---|---|
| 1 | p(HPMA-co-Ma-β-Ala-TT) | 10.7 | 40,000 | 1.06 |
| 2 | p(HPMA-co-Ma-β-Ala-DBCO) | 4.0 | 40,800 | 1.10 |
| 3 | p(HPMA-co-Ma-Acap-NHNH2) | 6.1 | 39,200 | 1.05 |
| 4 | p(HPMA-co-Ma-β-Ala-ATTO-488-co-Ma-β-Ala-DBCO) | 4.0 | 41,200 | 1.13 |
| Sample | Structure | Precursor | Peptide * wt% | Dye # wt% | Mw X g‧mol−1 | Đ X | DH XX nm |
|---|---|---|---|---|---|---|---|
| PEP | GKWMKLLKKILK-NH2 | N/A | 100 | 0 | N/A | N/A | N/A |
| F-PEP | ATTO-488-GKWMKLLKKILK-NH2 | N/A | 72.2 | 27.8 | N/A | N/A | N/A |
| P | p(HPMA) | N/A | 0 | 0 | 40,000 | 1.06 | 5.9 |
| P-PEP | p(HPMA-co-Ma-β-Ala-DBCO-azide-PEP) | 2 | 11.0 | 0 | 44,800 | 1.08 | 12.6 |
| F-P | p(HPMA-co-Ma-β-Ala-ATTO-488) | 2 | 0 | 2.0 | 40,000 | 1.06 | 8.2 |
| F-P-PEP | p(HPMA-co-Ma-β-Ala-ATTO-488-co-Ma-β-Ala-DBCO-azide-PEP) | 4 | 11.5 | 1.6 | 46,300 | 1.12 | n.d. |
| P-ValCit-PEP | p(HPMA-co-Ma-β-Ala-DBCO-ValCit-PEP) | 2 | 10.0 | 0 | 43,300 | 1.12 | 12.2 |
| P-LAAG-PEP | p(HPMA-co-Ma-β-Ala-DBCO-LAAG-PEP) | 2 | 12.8 | 0 | 42,000 | 1.08 | 11.2 |
| P-GFLG-PEP | p(HPMA-co-Ma-β-Ala-DBCO-GFLG-PEP) | 2 | 11.0 | 0 | 41,200 | 1.09 | 11.0 |
| P-Hyd-PEP | p(HPMA-co-Ma-Acap-NHN=levulinyl-PEP) | 3 | 14.8 | 0 | 45,500 | 1.18 | 12.6 |
| Bacterial Strain | PEP | P-PEP | P-ValCit-PEP | P-LAAG-PEP | P-GFLG-PEP | P-Hyd-PEP | F-PEP | F-P-PEP |
|---|---|---|---|---|---|---|---|---|
| S.aureus * | 10 | - | - | - | - | 12.5 | 72.9 | - |
| S. epidermidis * | 3.4 | 14.8 | - | 172.5 | 37–74.1 | 6.3 | 29.2 | 77.5 |
| A. baumanii † | 5 | 9.3 | 16.8 | 10.8–21.6 | 9.2 | 6.3 | 36.5 | 38.7 |
| E. coli † | 5 | 74.1 | 134 | 43.1 | 18.5 | 6.3 | 18.2 | 77.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pola, R.; Vícha, M.; Trousil, J.; Grosmanová, E.; Pechar, M.; Rumlerová, A.; Studenovský, M.; Kučerová, E.; Ulbrich, P.; Vokatá, B.; et al. Polymer-Antimicrobial Peptide Constructs with Tailored Drug-Release Behavior. Pharmaceutics 2023, 15, 406. https://doi.org/10.3390/pharmaceutics15020406
Pola R, Vícha M, Trousil J, Grosmanová E, Pechar M, Rumlerová A, Studenovský M, Kučerová E, Ulbrich P, Vokatá B, et al. Polymer-Antimicrobial Peptide Constructs with Tailored Drug-Release Behavior. Pharmaceutics. 2023; 15(2):406. https://doi.org/10.3390/pharmaceutics15020406
Chicago/Turabian StylePola, Robert, Matěj Vícha, Jiří Trousil, Eliška Grosmanová, Michal Pechar, Anna Rumlerová, Martin Studenovský, Emilie Kučerová, Pavel Ulbrich, Barbora Vokatá, and et al. 2023. "Polymer-Antimicrobial Peptide Constructs with Tailored Drug-Release Behavior" Pharmaceutics 15, no. 2: 406. https://doi.org/10.3390/pharmaceutics15020406