

Smart Poly(lactide)-b-poly(triethylene glycol methyl ether methacrylate) (PLA-b-PTEGMA) Block Copolymers: One-Pot Synthesis, Temperature Behavior, and Controlled Release of Paclitaxel

 and
and
Abstract
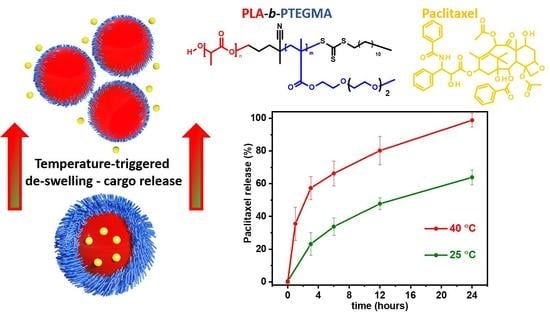
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Size Exclusion Chromatography (SEC)
2.3. NMR Spectroscopy
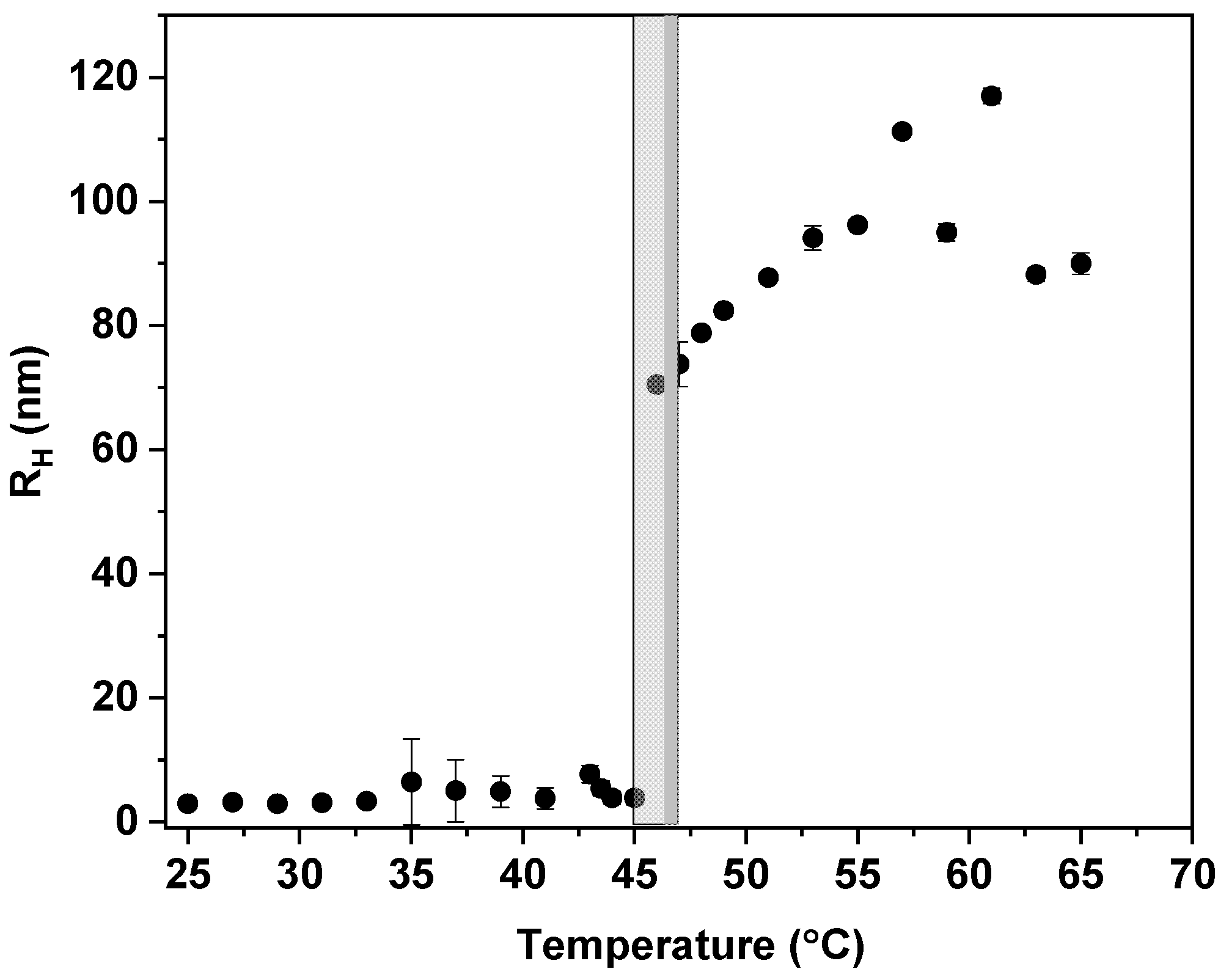
2.4. Dynamic Light Scattering (DLS)
2.5. Nanoparticles Preparation
2.6. Paclitaxel Loading
2.7. Drug Release Experiments
2.8. In Vitro Stability
2.9. Transmission Electron Microscopy (TEM)
3. Results and Discussion
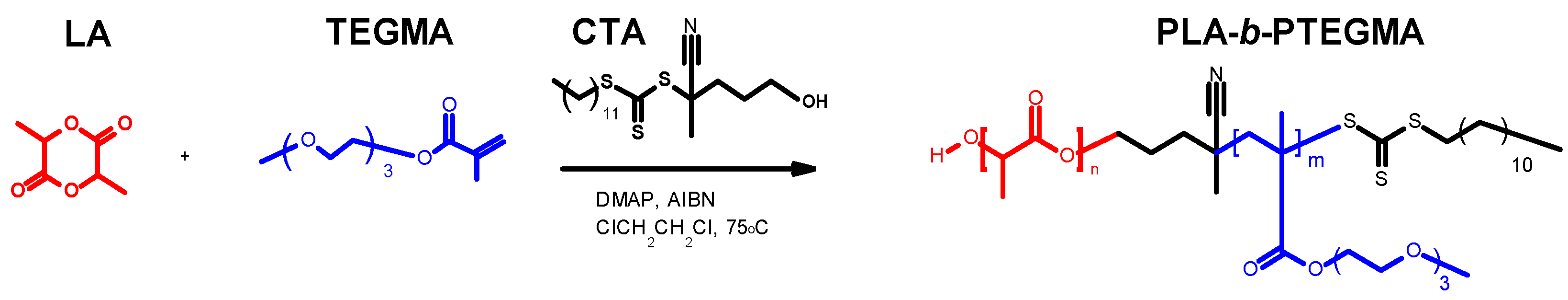
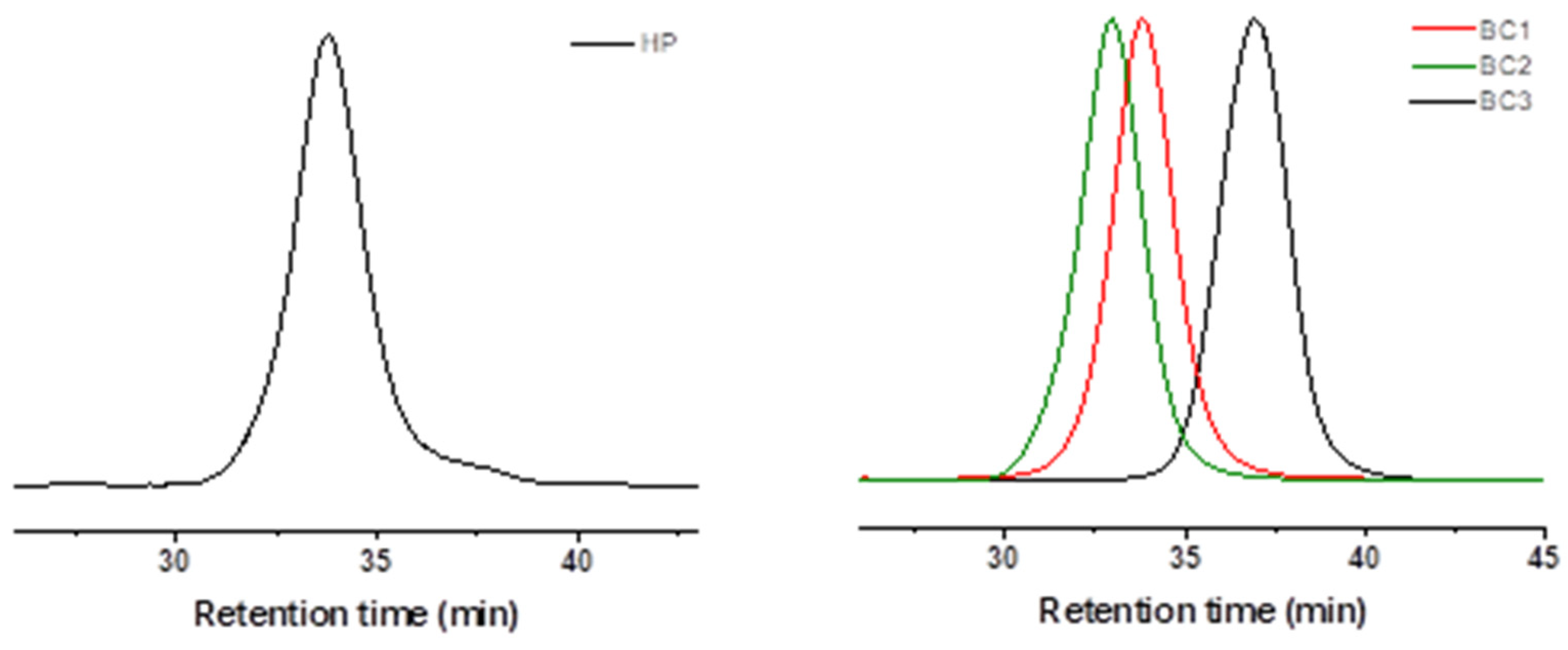
3.1. Synthesis of PLA-b-PTEGMA Block Copolymer via a One-Pot Protocol
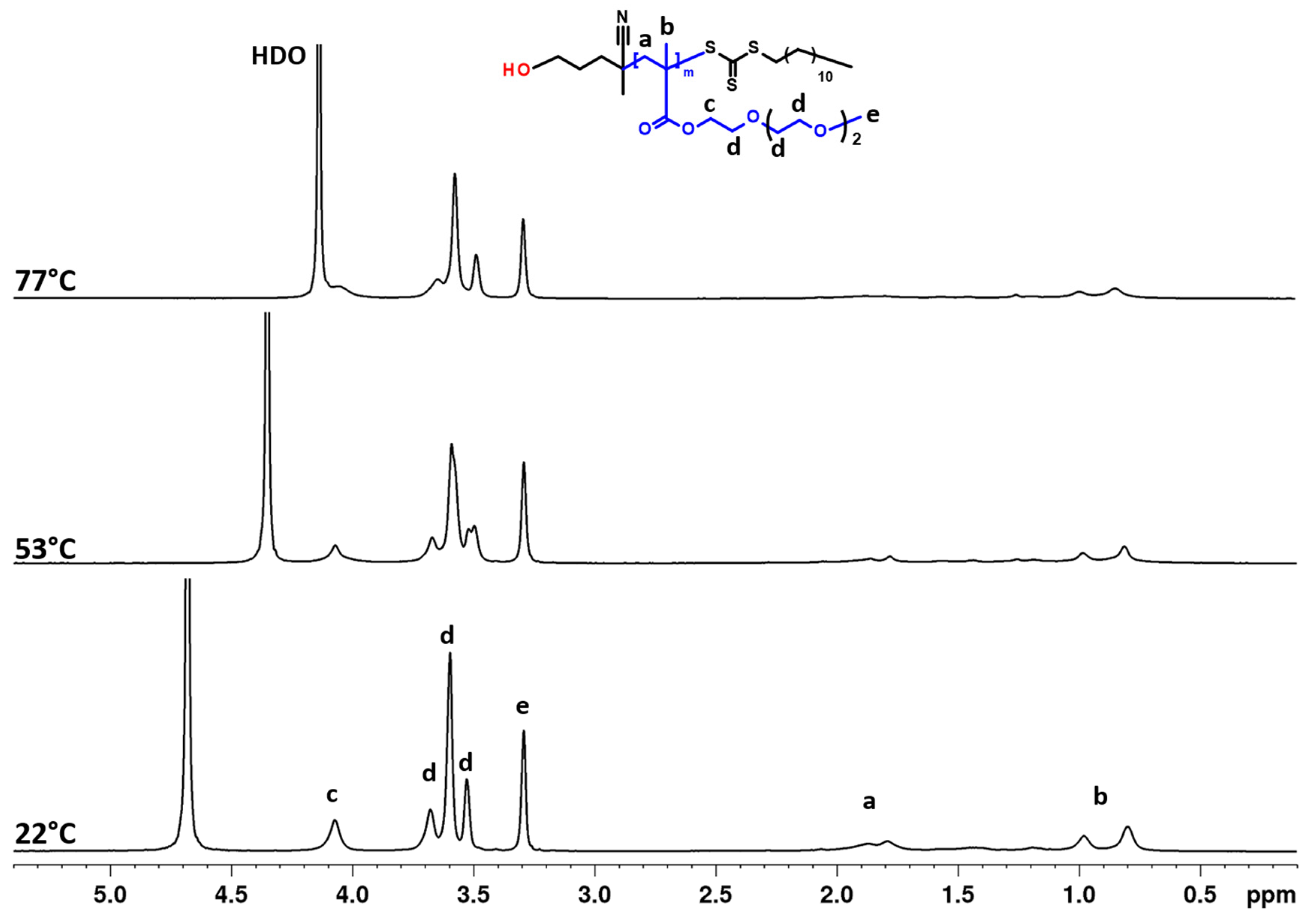
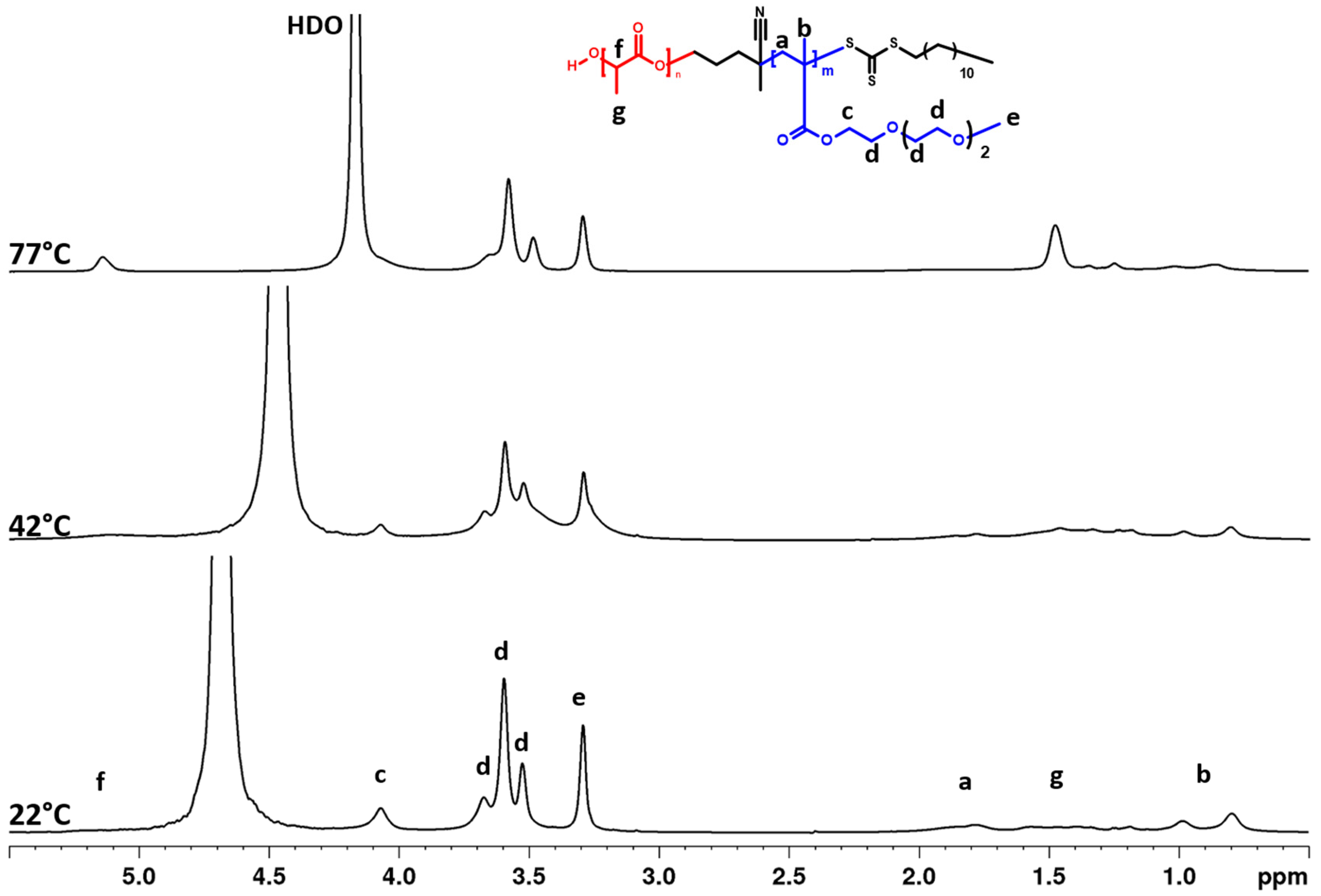
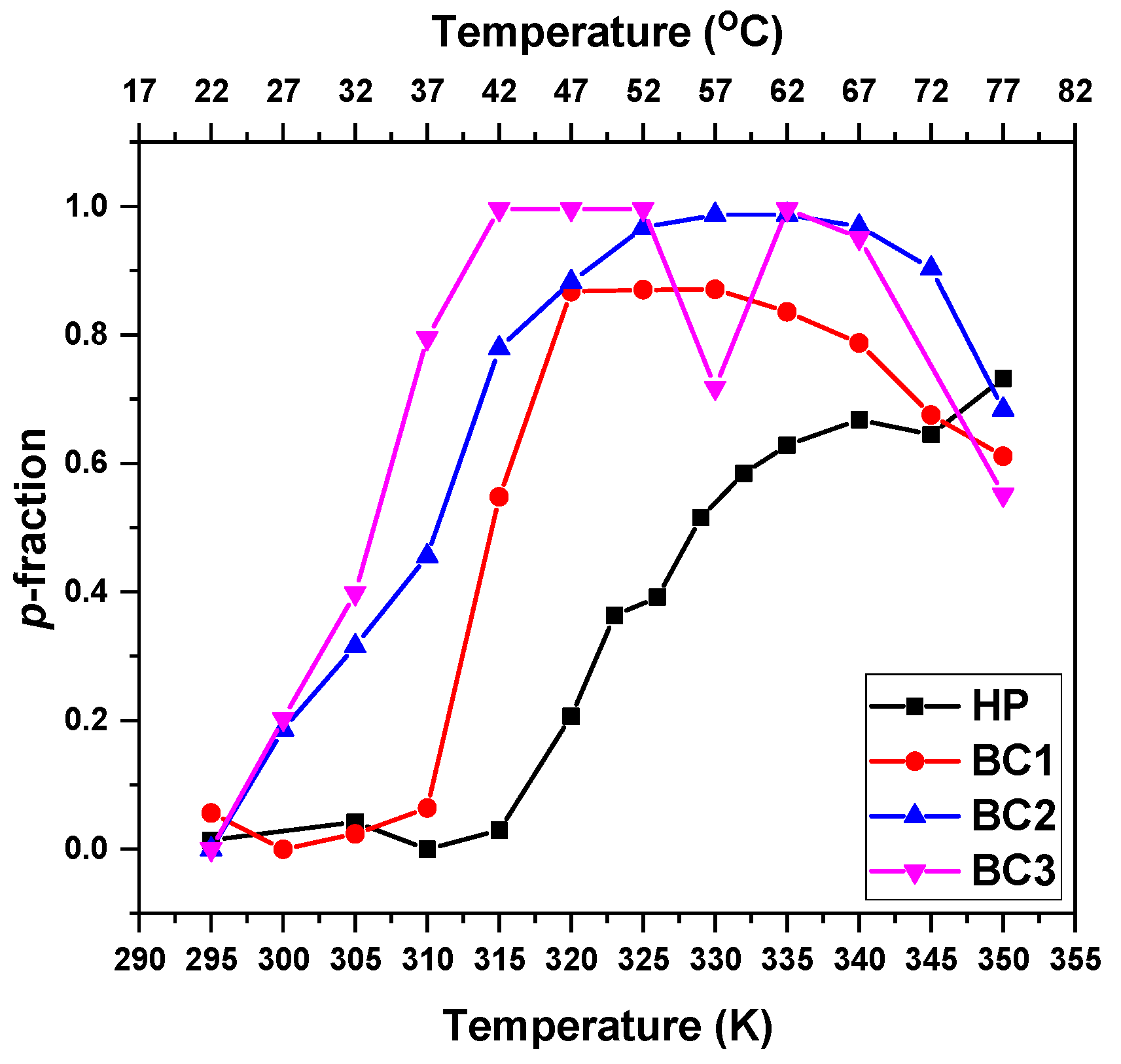
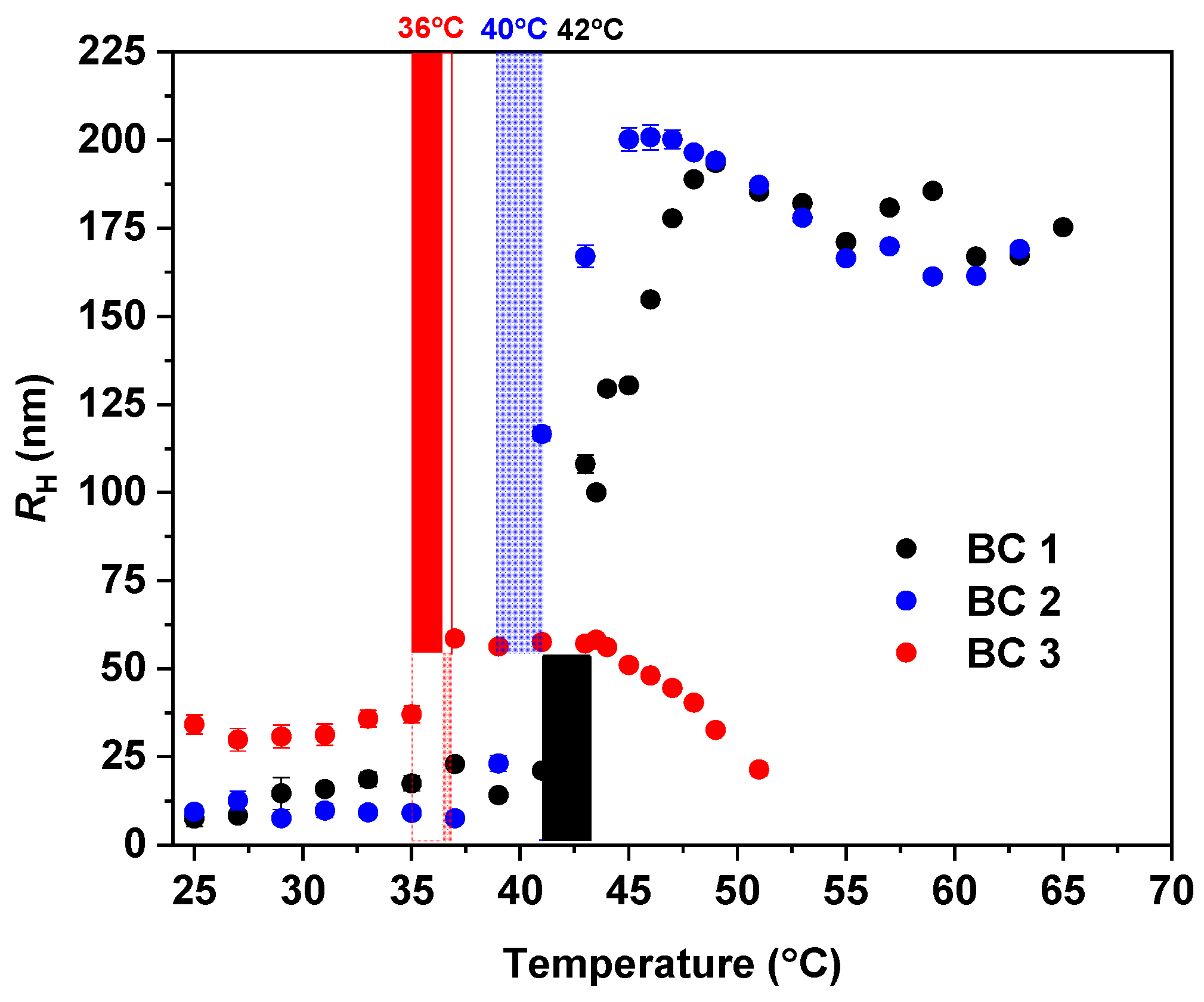
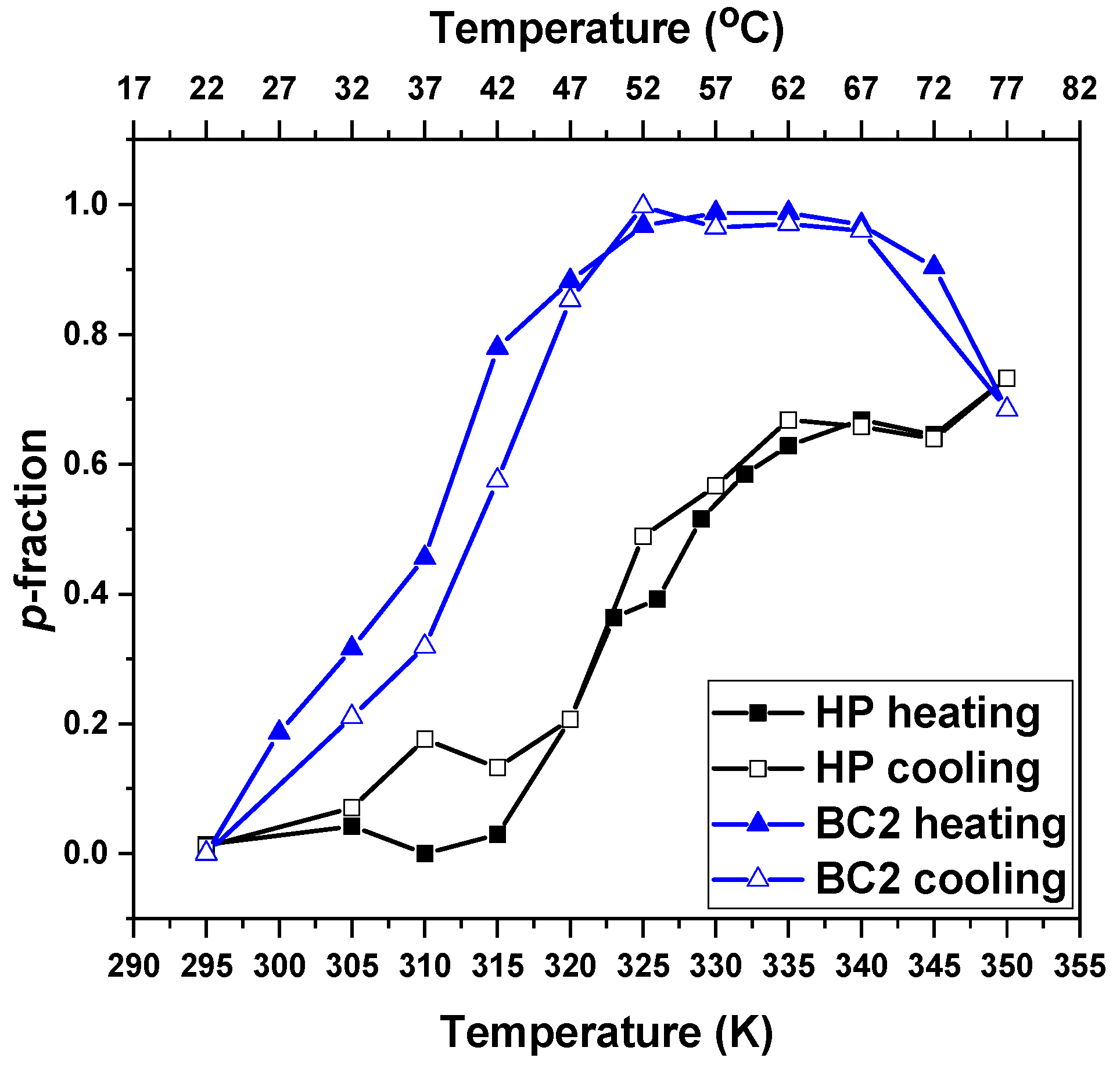
3.2. Temperature Behavior: DLS, 1H NMR Spectra, and Fraction p of Proton Units (Groups) with Reduced Mobility
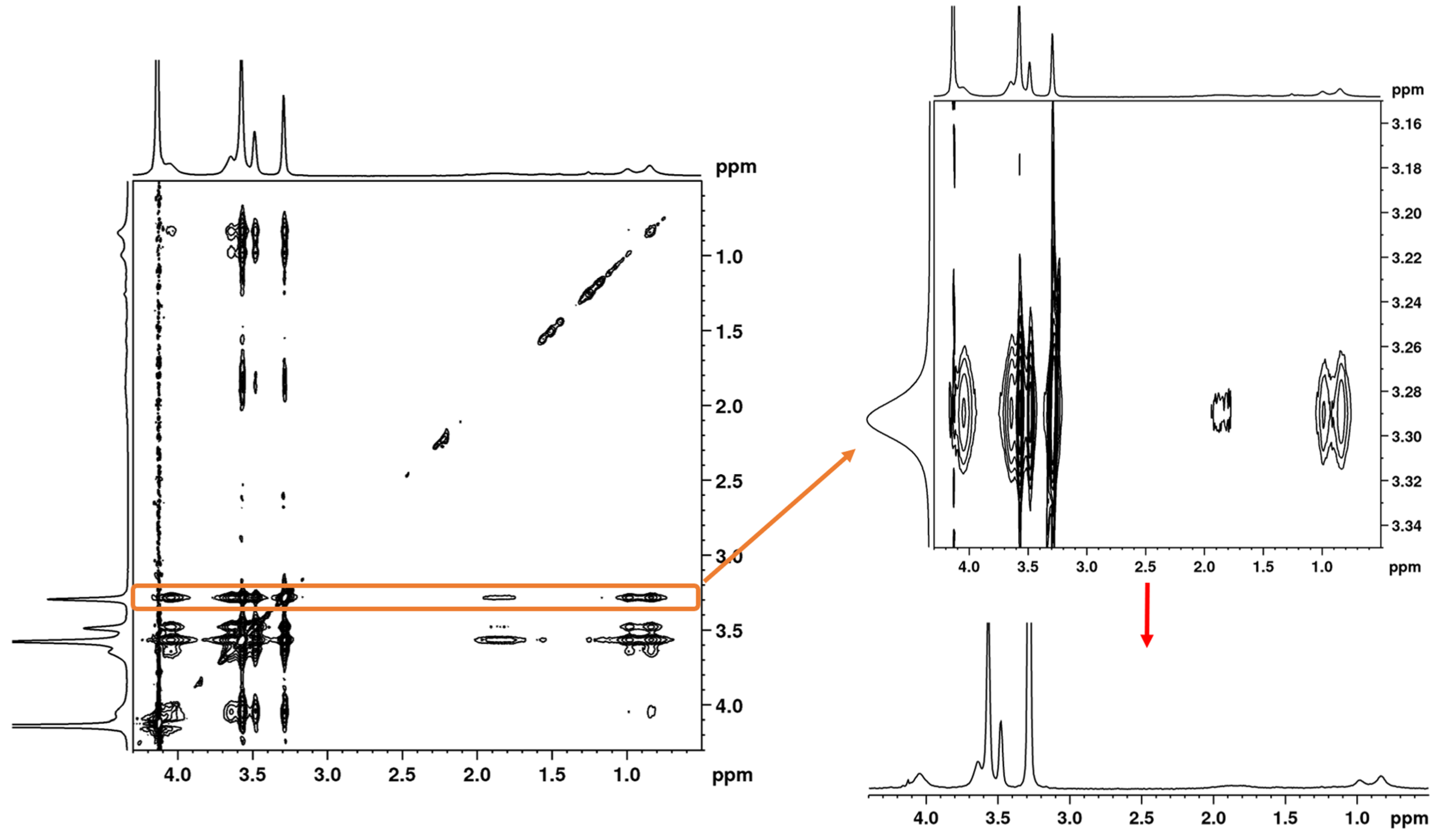
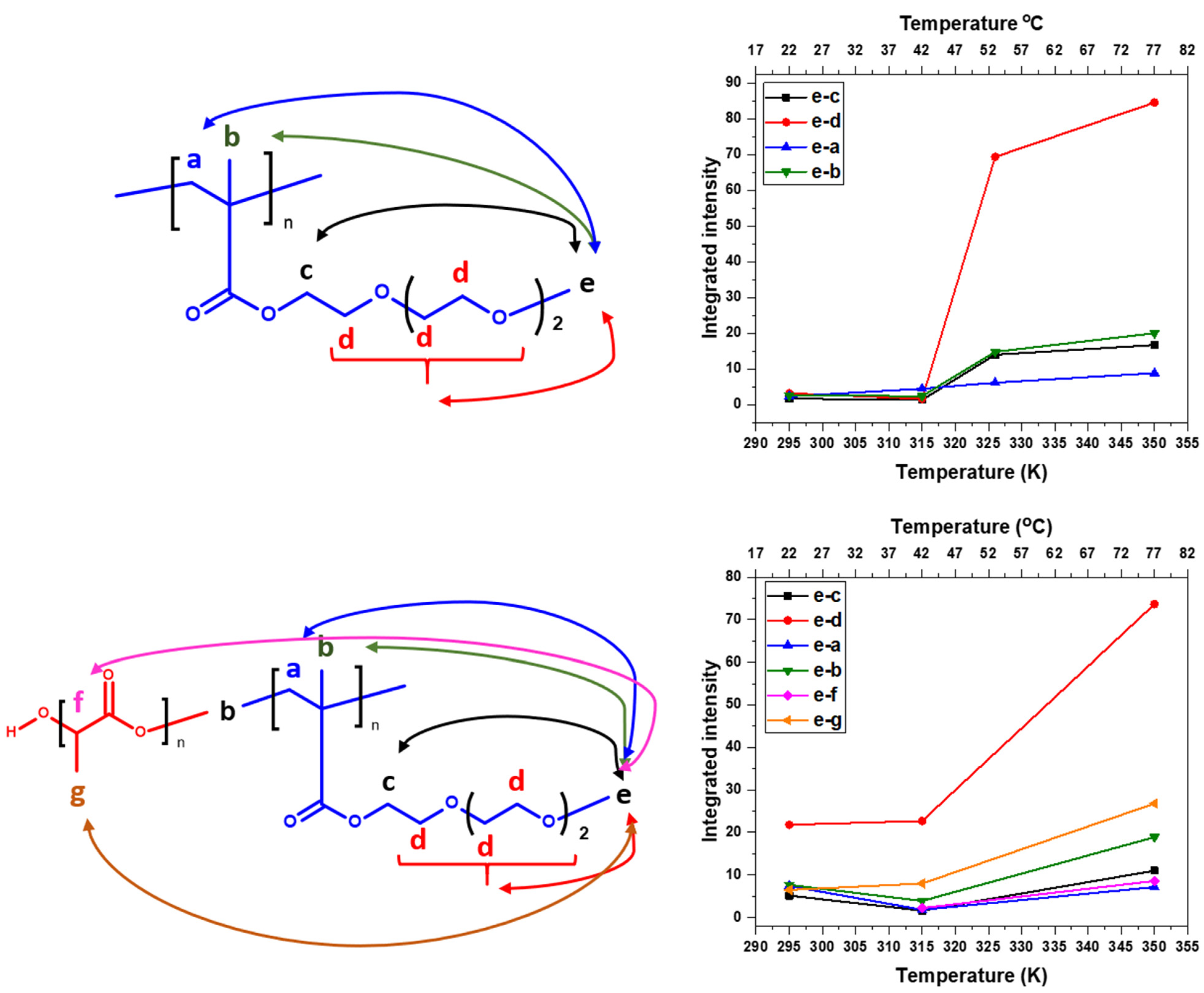
3.3. 2D 1H–1H NOESY NMR Spectra: Conformational Changes of Polymers
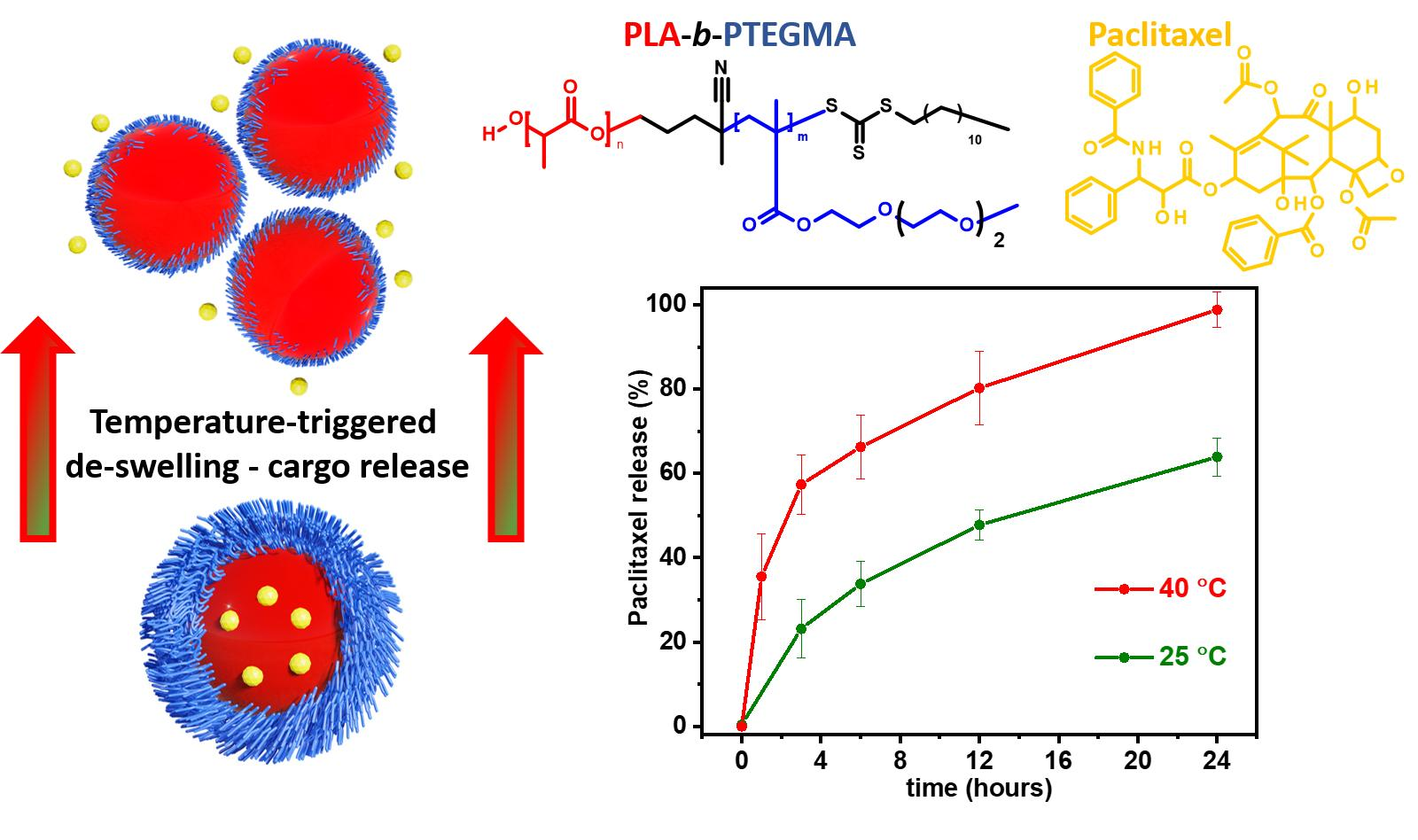
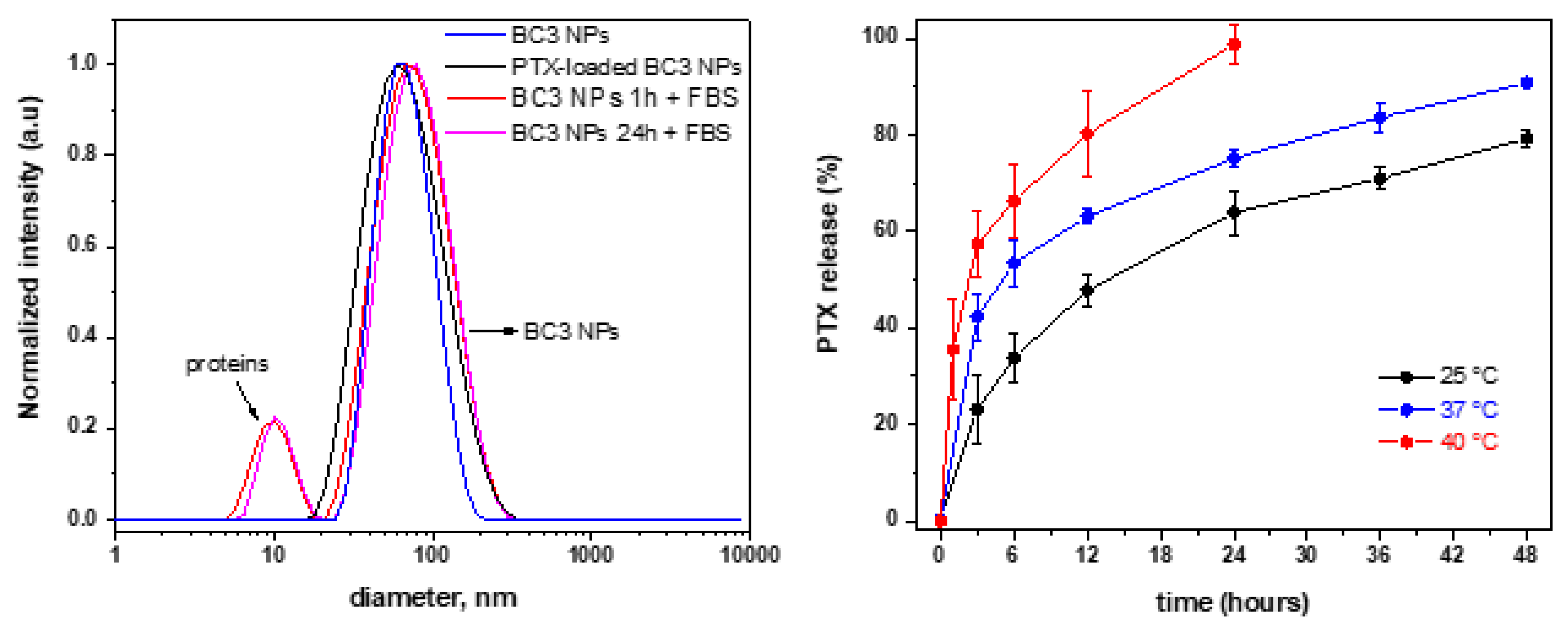
3.4. Paclitaxel-Loaded BC3 Nanoparticles and Paclitaxel Release
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ruiz, A.L.; Ramirez, A.; McEnnis, K. Single and Multiple Stimuli-Responsive Polymer Particles for Controlled Drug Delivery. Pharmaceutics 2022, 14, 421. [Google Scholar] [CrossRef] [PubMed]
- Urbánek, T.; Jäger, E.; Jäger, A.; Hrubý, M. Selectively biodegradable polyesters: Nature-inspired construction materials for future biomedical applications. Polymers 2019, 11, 1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, C.M.; Harris, M.; Choi, L.; Murali, V.P.; Guerra, F.D.; Jennings, J.A. Stimuli-responsive drug release from smart polymers. J. Funct. Biomater. 2019, 10, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.G.; De, P. pH responsive polymers with amino acids in the side chains and their potential applications. J. Appl. Polym. Sci. 2014, 131, 41084. [Google Scholar] [CrossRef]
- Saravanakumar, G.; Kim, J.; Kim, W.J. Reactive-Oxygen-Species-Responsive Drug Delivery Systems: Promises and Challenges. Adv. Sci. 2017, 4, 1600124. [Google Scholar] [CrossRef] [PubMed]
- Bordat, A.; Boissenot, T.; Nicolas, J.; Tsapis, N. Thermoresponsive polymer nanocarriers for biomedical applications. Adv. Drug Deliv. Rev. 2019, 138, 167–192. [Google Scholar] [CrossRef] [PubMed]
- Hruby, M.; Štěpánek, P.; Pánek, J.; Papadakis, C.M. Crosstalk between responsivities to various stimuli in multiresponsive polymers: Change in polymer chain and external environment polarity as the key factor. Colloid Polym. Sci. 2019, 297, 1383–1401. [Google Scholar] [CrossRef]
- Liu, F.; Urban, M.W. Recent advances and challenges in designing stimuli-responsive polymers. Prog. Polym. Sci. 2010, 35, 3–23. [Google Scholar] [CrossRef]
- Zarrintaj, P.; Jouyandeh, M.; Ganjali, M.R.; Hadavand, B.S.; Mozafari, M.; Sheiko, S.S.; Vatankhah-Varnoosfaderani, M.; Gutiérrez, T.J.; Saeb, M.R. Thermo-sensitive polymers in medicine: A review. Eur. Polym. J. 2019, 117, 402–423. [Google Scholar] [CrossRef]
- Manouras, T.; Vamvakaki, M. Field responsive materials: Photo-, electro-, magnetic- and ultrasound-sensitive polymers. Polym. Chem. 2017, 8, 74–96. [Google Scholar] [CrossRef]
- Sedlacek, O.; Hoogenboom, R. Drug Delivery Systems Based on Poly(2-Oxazoline)s and Poly(2-Oxazine)s. Adv. Ther. 2020, 3, 1900168. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Sanchez, R.J.P.; Fu, C.; Clayden-Zabik, R.; Peng, H.; Kempe, K.; Whittaker, A.K. Importance of Thermally Induced Aggregation on 19 F Magnetic Resonance Imaging of Perfluoropolyether-Based Comb-Shaped Poly(2-oxazoline)s. Biomacromolecules 2019, 20, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Sedlacek, O.; de la Rosa, V.R.; Hoogenboom, R. Poly(2-oxazoline)-protein conjugates. Eur. Polym. J. 2019, 120, 109246. [Google Scholar] [CrossRef] [Green Version]
- Schmaljohann, D. Thermo- and pH-responsive polymers in drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 1655–1670. [Google Scholar] [CrossRef]
- Ward, M.A.; Georgiou, T.K. Thermoresponsive polymers for biomedical applications. Polymers 2011, 3, 1215–1242. [Google Scholar] [CrossRef] [Green Version]
- Doberenz, F.; Zeng, K.; Willems, C.; Zhang, K.; Groth, T. Thermoresponsive polymers and their biomedical application in tissue engineering-A review. J. Mater. Chem. B 2020, 8, 607–628. [Google Scholar] [CrossRef]
- Wan, Z.; Zhang, P.; Liu, Y.; Lv, L.; Zhou, Y. Four-dimensional bioprinting: Current developments and applications in bone tissue engineering. Acta Biomater. 2020, 101, 26–42. [Google Scholar] [CrossRef]
- Aseyev, V.; Tenhu, H.; Winnik, F.M. Non-ionic Thermoresponsive Polymers in Water. Adv. Polym. Sci. 2010, 242, 29–89. [Google Scholar] [CrossRef] [Green Version]
- Halperin, A.; Kröger, M.; Winnik, F.M. Poly(N-isopropylacrylamide) Phase Diagrams: Fifty Years of Research. Angew. Chemie-Int. Ed. 2015, 54, 15342–15367. [Google Scholar] [CrossRef]
- Rusu, M.; Wohlrab, S.; Kuckling, D.; Möhwald, H.; Schönhoff, M. Coil-to-globule transition of PNIPAM graft copolymers with charged side chains: A1H and2H NMR and spin relaxation study. Macromolecules 2006, 39, 7358–7363. [Google Scholar] [CrossRef]
- Schild, H.G. Poly(N-isopropylacrylamide): Experiment, theory and application. Prog. Polym. Sci. 1992, 17, 163–249. [Google Scholar] [CrossRef]
- Spěváček, J.; Konefał, R.; Čadová, E. NMR Study of Thermoresponsive Block Copolymer in Aqueous Solution. Macromol. Chem. Phys. 2016, 217, 1370–1375. [Google Scholar] [CrossRef]
- Jana, S.; Uchman, M. Poly(2-oxazoline)-based stimulus-responsive (Co)polymers: An overview of their design, solution properties, surface-chemistries and applications. Prog. Polym. Sci. 2020, 106, 101252. [Google Scholar] [CrossRef]
- Hoogenboom, R.; Schlaad, H. Thermoresponsive poly(2-oxazoline)s, polypeptoids, and polypeptides. Polym. Chem. 2017, 8, 24–40. [Google Scholar] [CrossRef] [Green Version]
- Roy, D.; Brooks, W.L.A.; Sumerlin, B.S. New directions in thermoresponsive polymers. Chem. Soc. Rev. 2013, 42, 7214–7243. [Google Scholar] [CrossRef] [PubMed]
- Constantinou, A.P.; Tall, A.; Li, Q.; Georgiou, T.K. Liquid–liquid phase separation in aqueous solutions of poly(ethylene glycol) methacrylate homopolymers. J. Polym. Sci. 2022, 60, 188–198. [Google Scholar] [CrossRef]
- Li, Q.; Constantinou, A.P.; Georgiou, T.K. A library of thermoresponsive PEG-based methacrylate homopolymers: How do the molar mass and number of ethylene glycol groups affect the cloud point? J. Polym. Sci. 2021, 59, 230–239. [Google Scholar] [CrossRef]
- Liu, G.; Li, Y.; Yang, L.; Wei, Y.; Wang, X.; Wang, Z.; Tao, L. Cytotoxicity study of polyethylene glycol derivatives. RSC Adv. 2017, 7, 18252–18259. [Google Scholar] [CrossRef] [Green Version]
- Lutz, J.F. Thermo-switchable materials prepared using the OEGMA-platform. Adv. Mater. 2011, 23, 2237–2243. [Google Scholar] [CrossRef]
- Lutz, J.F.; Hoth, A. Preparation of ideal PEG analogues with a tunable thermosensitivity by controlled radical copolymerization of 2-(2-methoxyethoxy)ethyl methacrylate and oligo(ethylene glycol) methacrylate. Macromolecules 2006, 39, 893–896. [Google Scholar] [CrossRef]
- Han, S.; Hagiwara, M.; Ishizone, T. Synthesis of Thermally Sensitive Water-Soluble Polymethacrylates by Living Anionic Polymerizations of Oligo(ethylene glycol) Methyl Ether Methacrylates. Macromolecules 2003, 36, 8312–8319. [Google Scholar] [CrossRef]
- Szweda, D.; Szweda, R.; Dworak, A.; Trzebicka, B. Thermoresponsive poly[oligo(ethylene glycol) methacrylate]s and their bioconjugates-Synthesis and solution behavior. Polimery/Polymers 2017, 62, 298–310. [Google Scholar] [CrossRef]
- Yao, Z.L.; Tam, K.C. Temperature induced micellization and aggregation of biocompatible poly (oligo(ethylene glycol)methyl ether methacrylate) block copolymer analogs in aqueous solutions. Polymer 2012, 53, 3446–3453. [Google Scholar] [CrossRef]
- Zuppardi, F.; Chiacchio, F.R.; Sammarco, R.; Malinconico, M.; Gomez d’Ayala, G.; Cerruti, P. Fluorinated oligo(ethylene glycol) methacrylate-based copolymers: Tuning of self assembly properties and relationship with rheological behavior. Polymer 2017, 112, 169–179. [Google Scholar] [CrossRef]
- Guo, Y.; Dong, X.; Ruan, W.; Shang, Y.; Liu, H. A thermo-sensitive OEGMA-based polymer: Synthesis, characterization and interactions with surfactants in aqueous solutions with and without salt. Colloid Polym. Sci. 2017, 295, 327–340. [Google Scholar] [CrossRef]
- Maeda, Y.; Nakamura, T.; Ikeda, I. Hydration and phase behavior of poly(N-vinylcaprolactam) and poly(N-vinylpyrrolidone) in water. Macromolecules 2002, 35, 217–222. [Google Scholar] [CrossRef]
- Diab, C.; Akiyama, Y.; Kataoka, K.; Winnik, F.M. Microcalorimetric Study of the Temperature-Induced Phase Separation in Aqueous Solutions of Poly(2-isopropyl-2-oxazolines). Macromolecules 2004, 37, 2556–2562. [Google Scholar] [CrossRef]
- Maeda, Y. IR Spectroscopic Study on the Hydration and the Phase Transition of Poly(vinyl methyl ether) in Water. Langmuir 2001, 17, 1737–1742. [Google Scholar] [CrossRef]
- Martinez-Moro, M.; Jenczyk, J.; Giussi, J.M.; Jurga, S.; Moya, S.E. Kinetics of the thermal response of poly(N-isopropylacrylamide co methacrylic acid) hydrogel microparticles under different environmental stimuli: A time-lapse NMR study. J. Colloid Interface Sci. 2020, 580, 439–448. [Google Scholar] [CrossRef]
- Kadlubowski, S.; Matusiak, M.; Jenczyk, J.; Olejniczak, M.N.; Kozanecki, M.; Okrasa, L. Radiation-induced synthesis of thermo-sensitive, gradient hydrogels based on 2-(2-methoxyethoxy)ethyl methacrylate. Radiat. Phys. Chem. 2014, 100, 23–31. [Google Scholar] [CrossRef]
- Spěváček, J. NMR investigations of phase transition in aqueous polymer solutions and gels. Curr. Opin. Colloid Interface Sci. 2009, 14, 184–191. [Google Scholar] [CrossRef]
- Hofmann, C.H.; Schönhoff, M. Dynamics and distribution of aromatic model drugs in the phase transition of thermoreversible poly(N-isopropylacrylamide) in solution. Colloid Polym. Sci. 2012, 290, 689–698. [Google Scholar] [CrossRef]
- Kouřilová, H.; Šťastná, J.; Hanyková, L.; Sedláková, Z.; Spěváček, J. 1H NMR study of temperature-induced phase separation in solutions of poly(N-isopropylmethacrylamide-co-acrylamide) copolymers. Eur. Polym. J. 2010, 46, 1299–1306. [Google Scholar] [CrossRef]
- Zhang, C.; Peng, H.; Whittaker, A.K. NMR investigation of effect of dissolved salts on the thermoresponsive behavior of oligo(ethylene glycol)-methacrylate-based polymers. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 2375–2385. [Google Scholar] [CrossRef]
- Zhang, C.; Peng, H.; Li, W.; Liu, L.; Puttick, S.; Reid, J.; Bernardi, S.; Searles, D.J.; Zhang, A.; Whittaker, A.K. Conformation Transitions of Thermoresponsive Dendronized Polymers across the Lower Critical Solution Temperature. Macromolecules 2016, 49, 900–908. [Google Scholar] [CrossRef]
- Repasky, E.A.; Evans, S.S.; Dewhirst, M.W. Temperature Matters ! And Why It Should Matter to Tumor Immunologists. 2013, 1, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Gomes, I.P.; Duarte, J.A.; Maia, A.L.C.; Rubello, D.; Townsend, D.M.; de Barros, A.L.B.; Leite, E.A. Thermosensitive nanosystems associated with hyperthermia for cancer treatment. Pharmaceuticals 2019, 12, 171. [Google Scholar] [CrossRef] [Green Version]
- Jäger, A.; Gromadzki, D.; Jäger, E.; Giacomelli, F.C.; Kozlowska, A.; Kobera, L.; Brus, J.; Íhová, B.; El Fray, M.; Ulbrich, K.; et al. Novel “soft” biodegradable nanoparticles prepared from aliphatic based monomers as a potential drug delivery system. Soft Matter 2012, 8, 4343–4354. [Google Scholar] [CrossRef]
- Themistou, E.; Battaglia, G.; Armes, S.P. Facile synthesis of thiol-functionalized amphiphilic polylactide- methacrylic diblock copolymers. Polym. Chem. 2014, 5, 1405–1417. [Google Scholar] [CrossRef] [Green Version]
- García-Peñas, A.; Biswas, C.S.; Liang, W.; Wang, Y.; Yang, P.; Stadler, F.J. Effect of Hydrophobic Interactions on Lower Critical Solution Temperature for. Polymers 2019, 11, 991. [Google Scholar] [CrossRef] [Green Version]
- Konefał, R.; Spěváček, J.; Mužíková, G.; Laga, R. Thermoresponsive behavior of poly(DEGMA)-based copolymers. NMR and dynamic light scattering study of aqueous solutions. Eur. Polym. J. 2020, 124, 109488. [Google Scholar] [CrossRef]
- Zeng, F.; Tong, Z.; Feng, H.N.m.r. investigation of phase separation in poly(N-isopropyl acrylamide)/water solutions. Polymer 1997, 38, 5539–5544. [Google Scholar] [CrossRef]
- Spěváček, J.; Konefał, R.; Dybal, J.; Čadová, E.; Kovářová, J. Thermoresponsive behavior of block copolymers of PEO and PNIPAm with different architecture in aqueous solutions: A study by NMR, FTIR, DSC and quantum-chemical calculations. Eur. Polym. J. 2017, 94, 471–483. [Google Scholar] [CrossRef]
- Hanykova, L.; Radecki, M. NMR and thermodynamic study of phase transition in aqueous solutions of thermoresponsive amphiphilic polymer. Chem. Lett. 2012, 41, 1044–1046. [Google Scholar] [CrossRef] [Green Version]
- Šťastná, J.; Ivaniuzhenkov, V.; Hanyková, L. External Stimuli-Responsive Characteristics of Poly(N,N′-diethylacrylamide) Hydrogels: Effect of Double Network Structure. Gels 2022, 8, 586. [Google Scholar] [CrossRef]
- Konefał, R.; Černoch, P.; Konefał, M.; Spěváček, J. Temperature behavior of aqueous solutions of poly(2-oxazoline) homopolymer and block copolymers investigated by NMR spectroscopy and dynamic light scattering. Polymers 2020, 12, 1879. [Google Scholar] [CrossRef]
- Aubrecht, K.B.; Grubbs, R.B. Synthesis and characterization of thermoresponsive amphiphilic block copolymers incorporating a poly(ethylene oxide-stat-propylene oxide) block. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 5156–5167. [Google Scholar] [CrossRef]
- Riley, T.; Stolnik, S.; Heald, C.R.; Xiong, C.D.; Garnett, M.C.; Illum, L.; Davis, S.S.; Purkiss, S.C.; Barlow, R.J.; Gellert, P.R. Physicochemical evaluation of nanoparticles assembled from poly(lactic acid)-poly(ethylene glycol) (PLA-PEG) block copolymers as drug delivery vehicles. Langmuir 2001, 17, 3168–3174. [Google Scholar] [CrossRef]
- Konefał, R.; Spěváček, J.; Jäger, E.; Petrova, S. Thermoresponsive behaviour of terpolymers containing poly(ethylene oxide), poly(2-ethyl-2-oxazoline) and poly(ε-caprolactone) blocks in aqueous solutions: An NMR study. Colloid Polym. Sci. 2016, 294, 1717–1726. [Google Scholar] [CrossRef]
- Loukotová, L.; Bogomolova, A.; Konefal, R.; Špírková, M.; Štěpánek, P.; Hrubý, M. Hybrid κ-carrageenan-based polymers showing “schizophrenic” lower and upper critical solution temperatures and potassium responsiveness. Carbohydr. Polym. 2019, 210, 26–37. [Google Scholar] [CrossRef]
- Yamamoto, S.-I.; Pietrasik, J.; Matyjaszewski, K. The effect of structure on the thermoresponsive nature of well-defined poly(oligo(ethylene oxide) methacrylates) synthesized by ATRP. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 194–202. [Google Scholar] [CrossRef]
- Benoit, C.; Talitha, S.; David, F.; Michel, S.; Anna, S.J.; Rachel, A.V.; Patrice, W. Dual thermo- and light-responsive coumarin-based copolymers with programmable cloud points. Polym. Chem. 2017, 8, 4512–4519. [Google Scholar] [CrossRef]
- Weber, C.; Hoogenboom, R.; Schubert, U.S. Temperature responsive bio-compatible polymers based on poly(ethylene oxide) and poly(2-oxazoline)s. Prog. Polym. Sci. 2012, 37, 686–714. [Google Scholar] [CrossRef]
- Zhang, C.; Peng, H.; Puttick, S.; Reid, J.; Bernardi, S.; Searles, D.J.; Whittaker, A.K. Conformation of hydrophobically modified thermoresponsive poly(OEGMA-co-TFEA) across the LCST revealed by NMR and molecular dynamics studies. Macromolecules 2015, 48, 3310–3317. [Google Scholar] [CrossRef]
- Wang, F.; Porter, M.; Konstantopoulos, A.; Zhang, P.; Cui, H. Preclinical development of drug delivery systems for paclitaxel-based cancer chemotherapy. J. Control. Release 2017, 267, 100–118. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Kim, D.W.; Shim, Y.H.; Bang, J.S.; Oh, H.S.; Kim, S.W.; Seo, M.H. In vivo evaluation of polymeric micellar paclitaxel formulation: Toxicity and efficacy. J. Control. Release 2001, 72, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Salvioni, L.; Rizzuto, M.A.; Bertolini, J.A.; Pandolfi, L.; Colombo, M.; Prosperi, D. Thirty Years of Cancer Nanomedicine: Success, Frustration, and Hope. Cancers 2019, 11, 1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miguel, R.d.A.; Hirata, A.S.; Jimenez, P.C.; Lopes, L.B.; Costa-Lotufo, L.V. Beyond Formulation: Contributions of Nanotechnology for Translation of Anticancer Natural Products into New Drugs. Pharmaceutics 2022, 14, 1722. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update post COVID-19 vaccines. Bioeng. Transl. Med. 2021, 6, e10246. [Google Scholar] [CrossRef]
- Moore, T.L.; Rodriguez-Lorenzo, L.; Hirsch, V.; Balog, S.; Urban, D.; Jud, C.; Rothen-Rutishauser, B.; Lattuada, M.; Petri-Fink, A. Nanoparticle colloidal stability in cell culture media and impact on cellular interactions. Chem. Soc. Rev. 2015, 44, 6287–6305. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, F.A.; Albuquerque, L.J.C.; Riske, K.A.; Jäger, E.; Giacomelli, F.C. Outstanding protein-repellent feature of soft nanoparticles based on poly(N-(2-hydroxypropyl) methacrylamide) outer shells. J. Colloid Interface Sci. 2020, 574, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, F.C.; Stepánek, P.; Giacomelli, C.; Schmidt, V.; Jäger, E.; Jäger, A.; Ulbrich, K. PH-triggered block copolymer micelles based on a pH-responsive PDPA (poly[2-(diisopropylamino)ethyl methacrylate]) inner core and a PEO (poly(ethylene oxide)) outer shell as a potential tool for the cancer therapy. Soft Matter 2011, 7, 9316–9325. [Google Scholar] [CrossRef]
- Chan, J.M.; Zhang, L.; Yuet, K.P.; Liao, G.; Rhee, J.W.; Langer, R.; Farokhzad, O.C. PLGA-lecithin-PEG core-shell nanoparticles for controlled drug delivery. Biomaterials 2009, 30, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Jäger, E.; Venturini, C.G.; Poletto, F.S.; Colomé, L.M.; Pohlmann, J.P.U.; Bernardi, A.; Battastini, A.M.O.; Guterres, S.S.; Pohlman, A.R. Sustained release from lipid-core nanocapsules by varying the core viscosity and the particle surface area. J. Biomed. Nanotechnol. 2009, 5, 130–140. [Google Scholar] [CrossRef]
- Petrova, S.; Venturini, C.G.; Jäger, A.; Jäger, E.; Černoch, P.; Kereïche, S.; Kováčik, L.; Raška, I.; Štěpánek, P. Novel thermo-responsive double-hydrophilic and hydrophobic MPEO-b-PEtOx-b-PCL triblock terpolymers: Synthesis, characterization and self-assembly studies. Polymer 2015, 59, 215–225. [Google Scholar] [CrossRef]
- Petrova, S.; Venturini, C.G.; Jäger, A.; Jäger, E.; Hrubý, M.; Pavlova, E.; Štěpánek, P. Supramolecular self-assembly of novel thermo-responsive double-hydrophilic and hydrophobic Y-shaped [MPEO-b-PEtOx-b-(PCL)2] terpolymers. RSC Adv. 2015, 5, 62844–62854. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Moreno, P.; de Vicente, J.; Nardecchia, S.; Marchal, J.A.; Boulaiz, H. Thermo-sensitive nanomaterials: Recent advance in synthesis and biomedical applications. Nanomaterials 2018, 8, 935. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | LA/TEGMA Feed Ratio | Conv. a (%) LA/TEGMA | PLA/TEGMA NMR b | Mn, 1H NMR [g·mol−1] c | Mn, SEC d [g·mol−1] | Ð d |
|---|---|---|---|---|---|---|
| PTEGMA14.2K (HP) | 0/65 | 0/94 | 0/100 | 14,200 | 14,600 | 1.16 |
| PLA0.86K-b-PTEGMA14.5K (BC1) | 12/65 | 88/96 | 14/86 | 15,400 | 14,700 | 1.16 |
| PLA5.2K-b-PTEGMA9.5K (BC2) | 70/43 | 96/95 | 59/41 | 14,800 | 14,300 | 1.21 |
| PLA2.6K-b-PTEGMA2K (BC3) | 36/9 | 92/94 | 80/20 | 4600 | 6200 | 1.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lukáš Petrova, S.; Vragović, M.; Pavlova, E.; Černochová, Z.; Jäger, A.; Jäger, E.; Konefał, R. Smart Poly(lactide)-b-poly(triethylene glycol methyl ether methacrylate) (PLA-b-PTEGMA) Block Copolymers: One-Pot Synthesis, Temperature Behavior, and Controlled Release of Paclitaxel. Pharmaceutics 2023, 15, 1191. https://doi.org/10.3390/pharmaceutics15041191
Lukáš Petrova S, Vragović M, Pavlova E, Černochová Z, Jäger A, Jäger E, Konefał R. Smart Poly(lactide)-b-poly(triethylene glycol methyl ether methacrylate) (PLA-b-PTEGMA) Block Copolymers: One-Pot Synthesis, Temperature Behavior, and Controlled Release of Paclitaxel. Pharmaceutics. 2023; 15(4):1191. https://doi.org/10.3390/pharmaceutics15041191
Chicago/Turabian StyleLukáš Petrova, Svetlana, Martina Vragović, Ewa Pavlova, Zulfiya Černochová, Alessandro Jäger, Eliézer Jäger, and Rafał Konefał. 2023. "Smart Poly(lactide)-b-poly(triethylene glycol methyl ether methacrylate) (PLA-b-PTEGMA) Block Copolymers: One-Pot Synthesis, Temperature Behavior, and Controlled Release of Paclitaxel" Pharmaceutics 15, no. 4: 1191. https://doi.org/10.3390/pharmaceutics15041191