Opening lecture SL1 SL2 SL3 SL4 SL5 SL6 SL7 SL8 SL9 SL10 SL11 SL12 SL13 SL14 SL15 SL16 SL17 SL18 SL19 SL20 SL21
BIOINSPIRED POLYMERS: Where east meets west and North meets south
Helmut Ringsdorf
Institute of Organic Chemistry, University Mainz, Duesbergweg 10-14, 55099 Mainz, Germany
Tradition and innovation are two decisive aspects of Science. Tradition is the basis for it is the cumulation of wisdom in the body of knowledge. To know what a subject is all about and to control it creates self-confidence. Innovation is the adventure because with the challenge comes the risk of calling into question one's own scientific identity. Tradition and solid, successful work are honoured and admired. But persisting in it soon leads to tiresome routine and to research of yesterday: Science can only be justified by challenge and demands the willingness and ability to give up classical views in an attempt to discover new horizons.
The application of polymers in medicine has already reached dizzy heights. In the development of this modern field of biomedicine the Microsymposia in Prague have played for decades an essential role as an interdisciplinary forum where the East could meet the West and the South could meet the North. Being already "classical", now polymers in medicine show all advantages and disadvantages of age: Birthdays can be celebrated, facts and results are available, scientific harvest is abundant but where are the adventures, where is the future? Do you expect an answer?
The presently known genetic basis for most diseases, the detailed insight into the complex/cooperative processes of intracellular and transmembrane trafficking as well as all the information about angiogenesis and vascularization are opening essential perspectives for tissue, cell, and cell compartment specific drug delivery. Here both tradition and innovation are needed, here, at the borderline between different disciplines of science, adventures are waiting. Neither uncritical optimism nor obstructive pessimism are justified. Excitement and the courage to set out on new frontiers, and in particular the willingness and capability for close cooperation are basic prerequisites for the adventure of Science. All the knowledge is available – certainly spread out in different disciplines – we only have to learn to use it.
Pharmacological Profile of HPMA-Copolymers of Camptothecin
V.R. Caiolfa, M. Farao, E. Frigerio A, C. Pellizzoni A, M. Zamai, E. Pesenti, A. Suarato
Numerous clinical trials conducted with camptothecin (CPT) analogs have shown that these drugs, which are potent antitumor agents, require a prolonged schedule of administration given continuously at low doses to achieve therapeutic efficacy. We demonstrate here that conjugation of CPT to N-(2-hydroxypropyl)methacrylamide (HPMA) greatly increases the therapeutic index of the drug by slowly releasing CPT in the tumor mass. Soluble HPMA copolymers were synthesized to contain CPT (5 and 10 wt %). CPT was covalently linked at its -hydroxyl group to the polymers through two degradable spacers, -Gly-Phe-Leu-Gly- and -Gly-6-aminohexanoyl-Gly- (-Gly-C6-Gly-). In-vitro, CPT-conjugates were fairly resistant to hydrolysis in plasma as in buffer at neutral pH (0.2 - 0.4 % free CPT /hr), while elastase and cysteine-proteases were able to release the active drug. Plasma levels of free CPT in mice after intravenous administration of CPT-copolymers were ~5 fold lower than those observed at the highest tolerated dose of CPT administered in classical vehicles. In contrast with native CPT, which was highly toxic by the intravenous route, CPT-conjugates induced complete tumor remission in nude mice with human MX1 mammary and A2780 ovarian carcinomas and M14 melanoma subcutaneous xenografts. The improved pharmacological profile of CPT-conjugates was also confirmed in A549 NSCL in xenografts with no sign of toxicity. In particular, HPMA-Gly-C6-Gly-CPT, at 10% CPT content, showed an outstanding antitumor activity also against orthotopic HT29 human colon carcinoma xenografts, with complete remission of the primary tumor and distant metastasis. The latter conjugate is presently undergoing preliminary clinical trials as the first HPMA copolymer of camptothecin.
SL2
SL3
MECHANISMS OF ANTICANCER ACTION OF HPMA COPOLYMER-BOUND DOXORUBICIN
T. MINKO, P. KOPEČKOVÁ, J. KOPEČEK
Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, 30 S 2000 E Rm. 301, Salt Lake City, Utah 84112, U.S.A.
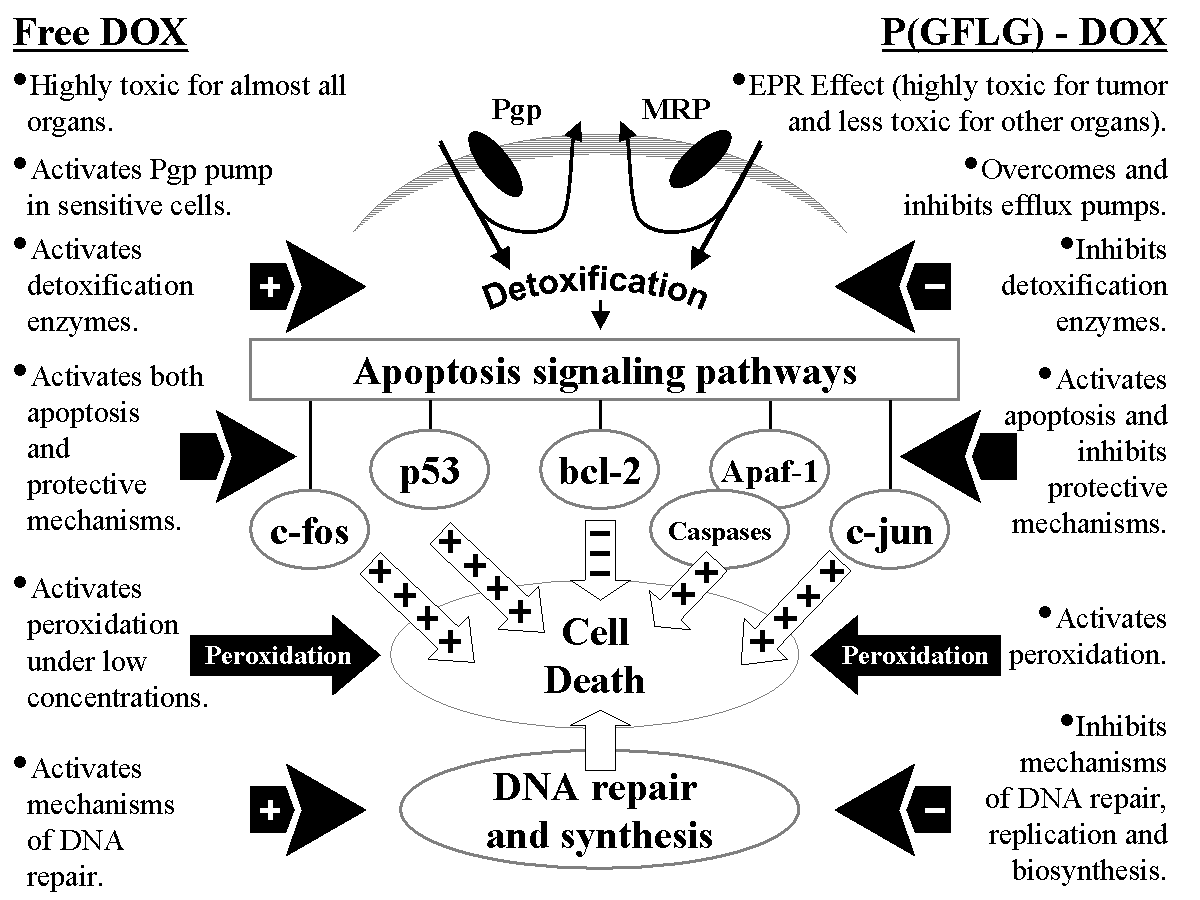
It was found that polymer-bound doxorubicin demonstrated higher anticancer activity when compared to free doxorubicin. This phenomenon was explained by the following mechanisms of its anticancer action: preferential accumulation in tumors; internalization in membrane-limited organelles; ability to overcome existing multidrug resistance and to not induce it de novo; high intracellular toxicity and inhibition of detoxification enzymes; cell death induction by the activation of specific signaling pathways; triggering of caspase activation cascades (Fig.1).
DESIGN OF SYNTHETIC POLYMERS TO ENHANCE INTRACELLULAR TRAFFICKING OF DNA
ALLAN HOFFMAN, CHUCK CHEUNG, CHANTAL LACKEY, NIREN MURTHY, PATRICK STAYTON (Bioengineering Dept.), THEMIS KYRIAKIDES, PAUL BORNSTEIN (Biochemistry Dept.), NELSON FAUSTO, JEAN CAMPBELL (Pathology Dept.), OLLIE PRESS (School of Medicine)
Univ. of Washington, Box 352255, Seattle, WA
One of the key steps in non-viral gene therapy or antisense therapy is delivery of the plasmid DNA or antisense oligonucleotides from the endosome to the cytoplasm, in order to avoid their degradation by lysosomal DNases. We are designing and synthesizing various pH-sensitive polymers and copolymers that are non-disruptive to cell membranes at physiologic pH, but become disruptive to endosomal membranes at the low pHs that exist within those vesicles. The abilities of these pH-sensitive homopolymers and copolymers to disrupt lipid bilayer membranes have been assessed in vitro by measuring their hemolytic activities at different pHs. We have then incorporated the most promising polymers into non-viral DNA delivery formulations and tested their ability to enhance the transfection of cells in culture and in vivo, using both cationic lipid and polycationic carrier systems. Recent results show significant enhancement of transfection by our polymers, both in vitro and in vivo. These results will be described and discussed.
SL6
NOVEL ASPECTS OF ORAL DRUG DELIVERY THROUGH POLYMERIC HYDROGELS
N.A. PLATE, L.I. VALUEV
Institute of Petrochemical Synthesis, Russian Academy of Sciences,
Leninsky prospect, 29, 117912 Moscow, Russia
A new approach to overcome the degradation of protein drugs by digestive enzymes and its targeting to the blood through digestive apparatus was developed. The approach is based on the immobilization of drugs into the polymeric hydrogel which is modified with polysaccharide and inhibitor of proteolytic enzymes. The mechanism of physiological effect of immobilized drugs involves the inhibition of drugs proteolysis due to neutralization of proteolytic enzymes by inhibitor and increase in the rate of drug diffusion from the particles of hydrogel through the membrane of small intestine. The latter is associated with a specific reaction between the polysaccharide chemically bound with hydrogel and lectin contained in the mucosal membrane of intestine. This binding leads to the increase in the local concentration of drug directly in the musocal membrane and, as a result, the rate of its diffusion to the blood stream is increased. As protein drugs calcitonin, glucagon, insulin and growth hormone were used. The effect of orally given immobilized preparations is in average not below 50 % of subcutaneously injected drugs in equal dosages.
New hydrogels on the base of copolymers of acrylamide with N-(2-D-glucose)acrylamide (GAA) crosslinked by Concanavalin A (Con A) were synthesized. When studying the reaction of these hydrogels with glucose solutions of various concentrations, it was found that there exists a threshold concentration, below which no detectable changes in the hydrogel are observed. At concentrations of glucose higher then the threshold concentration, hydrogel quickly transforms into a soluble state. This implies that the [Con A-GAA] complex decomposes to form a [Con A-glucose] complex and water-soluble GAA-acrylamide copolymer. The higher the content of GAA units in the copolymer, the higher the threshold concentration. Thus, the prepared hydrogels are glucose-sensitive systems capable of releasing the containing compounds, for example insulin.
SL8
SL9
TREATMENT OF INTRAOCULAR DISEASES WITH POLY(ORTHO ESTER)-BASED DRUG DELIVERY SYSTEMS
S. EINMAHL1, F. BEHAR-COHEN2, C. TABATABAY1, R. GURNY1.
1Department of Pharmaceutics and Biopharmaceutics, School of Pharmacy, University of Geneva, Quai Ernest-Ansermet 30, CH-1211 Geneva 4, Switzerland.
2Department of Ophthalmology, Hôtel-Dieu Hospital, Place du Parvis de Notre-Dame 1, F-75004 Paris, France.
Drug delivery to the posterior segment of the eye remains up to date a major challenge. Conventional drug administration to the eye, i.e. topical instillation, or systemic administration, do not provide sufficient drug levels for treating intraocular disorders, owing to the anatomical and physiological barriers that exist in the eye. The only way to attain therapeutic drug concentrations is by direct intraocular injection. Unfortunately, successful treatment requires multiple injections to maintain therapeutic drug concentrations in the eye for a desired period of time. Additionally, frequent intraocular injections can cause severe complications. Researchers have thus been encouraged to develop new systems for unique administration of drugs that would provide therapeutic drug levels to the posterior segment over an extended period of time.
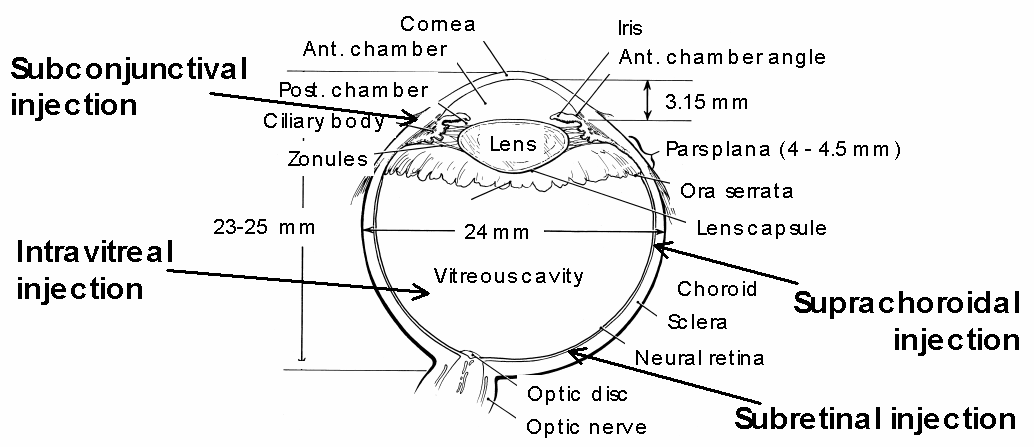
Figure 1: Scheme of a human eye showing the intraocular sites amenable to sustained drug delivery.
An injectable, bioerodible, third-generation poly(ortho ester) (POE) carrier has been developed for sustained drug delivery in ocular pathologies such as glaucoma filtering surgery failure, proliferative vitreoretinopathy, or age-related macular degeneration. The main advantage of using bioerodible polymers for drug delivery during ophthalmic surgery is that there is no need to remove the device once the drug released.
This presentation describes the intraocular application of POE-based delivery systems: a first part will present the intraocular biocompatibility assessment, and the second part will focus on the clinical results obtained with an animal model of glaucoma filtering surgery.
Supported by the Swiss National Science Foundation grant #3200-056750.99/1.
Tailor-made biomimetic random copolymers for medical applications
C. Gervelas, T. Avramoglou, J. Jozefonvicz, M. Jozefowicz
Laboratoire de Recherches sur les macromolécules, CNRS UMR 7040, Institut Galilée, Université Paris 13, 99 avenue J.B. Clément, 93430 Villetaneuse, France
Biospecific copolymers have been synthesized by random substitution of a preformed polymer with suitable chemical groups or random copolymerization of suitable functional monomers. Such polymers contain arrangements of the chemical functions that mimic natural biospecific sites. The probability of occurrence of such arrangements will depend on the average composition of the copolymer. According to this principle, two examples of ”bioactive copolymers” will be presented.
Some dextran derivatives termed dextran methylcarboxylic benzylamide (DMCB) exhibit an inhibitor effect on the growth of human breast cancer cell lines. The inhibitory capacity on cell growth depends on the benzylamide content. The active dextran derivatives exert their inhibitory effect on the cell proliferation of pretumoral and tumoral human mammary epithelial cell lines (HBL 100 and MCF-7) by interfering with specific autocrine and paracrine growth factors. Moreover, some of these derivatives were found to have a large antitumoral activity on malignant human melanoma 1205LU. In vivo preliminary investigations have started to test the antitumoral activity in athymic mice.
Moreover, the adhesion of Staphylococcus aureus onto biospecific random polystyrene derivatives carrying sulfonate and carboxylate groups is hindered in a composition dependant way. Concurrently, polyvinyl chloride and acrylic based random copolymers carrying the same chemical groups exhibit a similar effect. In addition, a correlation between the bacterial adhesion and proliferation has been evidence. As a consequence, biospecific random copolymers may be endowed with both bacteriophobic and bacteriostatic activities with regard to Staphylococcus aureus, one of the principal microorganisms involved in biomaterial-centered infections.
Modulated Insulin Delivery using Phase-Reversible Glucose-Sensitive Hydrogels
J.J. Kim, K. Park
Purdue University, Departments of Pharmaceutics and Biomedical Engineering, West Lafayette, IN 47907, U.S.A.
It is highly desirable to develop a simple, continuous, and non-invasive insulin delivery system mimicking physiological insulin release for the control of hyperglycemia and prevention of the resulting complications. An ideal insulin delivery system would have the capability to sense the glucose level in the blood and release insulin at an appropriate rate corresponding to the glucose level. We have prepared and characterized hydrogels that undergo reversible gel-sol phase transition in response to the changes of glucose concentrations. Glucose-containing polymers were prepared and characterized for allyl glucose content, monomer reactivity ratio, and molecular weight. Con A conjugated with monomethoxy poly(ethylene glycol) showed increased solubility and stability in aqueous solution. Modified Con A could form hydrogels with glucose-containing polymers. The phase diagrams for the gel formation between Con A or modified Con A and glucose-containing polymers were constructed. The mixture solutions containing modified Con A resulted in a clear gel and a sol, while Con A solutions with the copolymers resulted in various phases such as a clear gel, a turbid gel, precipitates, and a sol. The hydrogels composed of modified Con A underwent phase transition to sol in the presence of free glucose in the surrounding environment. Viscosity of the hydrogels significantly decreased by the addition of glucose to the hydrogels. The reversible phase transition between a gel and a sol was verified in response to the changes of glucose concentration over 30 days at room temperature. Insulin release study was performed using three kinds of insulin delivery systems such as membrane, matrix, and erodible matrix system. The glucose-sensitive hydrogels controlled the release rate of insulin as a function of glucose concentration.
Selective Energy Depletion and Sensitization of Multiple Drug Resistant Cancer Cells by Pluronic Block Copolymers
A.V. Kabanova, E.V. Batrakovaa, S. Lia, V.Yu. Alakhovb
aCollege of Pharmacy, 986025 Nebraska Medical Center, Omaha, NE 6819, USA
bSupratek Pharma Inc., 513 blvd. des Prairies, Case Post. 100, Laval, PQ, Canada H7N 4Z3
Poly(ethylene oxide)-poly(propylene oxide) block copolymers (Pluronic) have recently been used in formulations to treat resistant cancers. Pluronic "hypersensitize" multidrug resistant (MDR) cells, resulting in an increase in the cytotoxic activity of anthracyclines by 2 to 3 orders of magnitude. MDR cells overexpress ATP-binding proteins such as P-glycoprotein (Pgp) and multidrug resistance-associated proteins (MRP) that pump drugs out of a cell. Energy-dependent processes associated with the above drug resistance mechanisms impose higher energy requirements in MDR cells, which render these cells more sensitive to energy depleting agents than the wild type cells that have lower energy requirements. This paper for the first time demonstrates that Pluronic P85 induces a major decrease in ATP levels selectively in MDR cells, while non-MDR cells do not respond to this block copolymer. It further shows that energy depletion induced by Pluronic is responsible for the potentiation of the cytotoxic effects of anthracyclines in MDR cells.
biodegradable graft polyesters based on poly(lactide-co-glycolide) and pval for oral vaccine delivery
t. kissel, t. jung, a. breitenbach, w.kamm
Department of Pharmaceutics, Philipps-University Marburg, Ketzerbach 63,
35032 Marburg, Germany
A major challenge in oral drug delivery is to find suitable carrier systems for hydrophilic macromolecular drugs. We investigated poly[2-(sulfobutyl)vinyl alcohol]-graft-poly(lactide-co-glycolide), SB-PVAL-g-PLG, branched polyesters, combining a modified three-dimensional architecture, increased hydrophilicity of poly(lactide-co-glycolide) (PLG) and charged groups in a single polymer. These polymers allow preparation of nanoparticles (NP) with defined surface properties even without the use of surfactants in the water phase.
The polyelectrolyte backbones were obtained by reacting poly(vinyl alcohol) (PVAL) with charge-containing pendent groups. Biodegradable comb PLGs were synthesized by ring-opening melt polymerization of lactide and glycolide in the presence of different polyols. The polymers were characterized by nuclear magnetic resonance, infrared spectroscopy, differential scanning calorimetry, elemental analysis and measurement of intrinsic viscosity. NP, as colloidal carriers for tetanus toxoid, were prepared by controlled precipitation and investigated by surface NMR spectra as a function of polymer composition, surface charge and hydrophobicity. In the case of NP prepared from negatively charged polyesters, a core-corona-like NP structure with an inner polyester core and an outer charged groups containing coating seems to be likely. NP of different sizes, prepared from SB-PVAL-g-PLG, were loaded with tetanus toxoid by adsorption and applied to Balb/c mice for immunization by oral, nasal and ip application. Both p.o. and i.n. application significantly increased serum titres of IgA. Small (100 nm) NP yielded higher IgG and IgA titres after oral and especially after nasal administration than larger NP. Therefore, antigen-loaded NP prepared from SB-PVAL-g-PLG show considerable poential as a vaccine delivery system for mucosal immunization.
Oligopeptides as Potential Insulin Drug Delivery System
Raphael M. Ottenbrite, Rui Zhao and Mamoru Haratake
Dept of Chemistry, Virginia Commonwealth University
Richmond, VA USA 23284-2006
One of the primary interests in the pharmaceutical industry is the oral delivery of naturally occurring protein drugs. Insulin is one of the high priority drugs in this category due to the large number of diabetics throughout the world. Based on the success reported for proteinoid microspheres for the delivery of many natural occurring macromolecular drugs, we synthesized and characterized several quaternary oligopeptides of naturally occurring amino acids as potential oral drug delivery systems. Previously we reported the aggregation behavior of these oligopeptides and their interaction with heparin.
The concept for increased absorption of a drug is hydrophobize and to convert it to a molten form which facilitate uptake through the lower GI trac. Insulin is a globular protein with discrete secondary structures with alpha helix, beta turn and beta strands. Under certain conditions, insulin also self-aggregates to form dimmers, trimmers and hexamers. The effects of two hydrophobic tetrapeptide systems on the structure of insulin was studied by circular dichroism . The (CD) spectrum of insulin has two major bands, one at 208 nm and the other at 222nm as well as a minor peak at 273. These three CD bands have been assigned to the alpha and beta structures and the formation of higher self-aggregates, respectively. The addition of pEELL (3mg/mL) to an insulin solution (100 uM) resulted in the attenuation of the at 209 and 270 nm and enhancement of the band at 222 nm. The addition of 5mg/L of this tetrapeptide caused an enhancement and a shift of the 222 peak to 225. This was interpreted as an increase in the number of insulin molescules in an aggregated form. Conversely, the addition of pEEFF seams to have changed the alpha and beta structure indicating denaturation to the globular form. The aliphatic hydrophobic tetrapeptide pEELL behavior was significantly different from that of the aromatic hydrophobic tetrapeptide pEEFF. This phenomena was also observed for these tetrapeptides with heparin.
Assuming that these differences were due to the association of the tetrapeptides with insulin, isothermal titrations were conducted to measure the thermodynamic binding parameters. The heat capacity change when negative is a good indicator that hydrophobic interactions are occurring and positive hydrophobic actions are being perturbed.
POLYMERIC ORGANOIRON COMPOUNDS AS PRO-DRUGS IN CANCER RESEARCH
E.W. NEUSE
Department of Chemistry, University of the Witwatersrand, WITS 2050, South Africa
Cancerous diseases place a heavy burden on national health systems worldwide. Chemotherapy has proved a powerful weapon against neoplasias. However, despite appreciable progress in the field, overall rates of remission and cure by chemotherapy remain unduly low, largely because of toxic side effects and the development of drug resistance. The technology of polymer-drug conjugation involving reversible drug binding to water-soluble macromolecular carriers, first rationally articulated by Ringsdorf1, has since emerged as an advanced strategy designed to overcome the pharmacological deficiencies of todays clinically administered antineoplastic agents. The conjugate will act as a pro-drug, transporting the drug species in an inactive form to, and into, the target cells, where the active agent is hydrolytically or enzymatically released for pharmacological action. The present communication deals with polymer-ferrocene conjugates, a class of macromolecular organoiron compounds in which the active drug species is a derivative of the di-η5-cyclopentadienyliron(II) complex, ferrocene. The significant antiproliferative properties of certain monomeric ferrocene derivatives observed in previous work2 provided the driving force for a comprehensive synthetic program in this laboratory which has resulted in the development of polymeric ferrocene conjugates based on water-soluble and biodegradable polyamide carriers of various structures interconnected with the ferrocene unit via biofissionable amide or ester links. Preliminary in vitro screens performed on selected conjugates show highly promising antiproliferative activity against HeLa and LNCaP human cancer cell lines, well on a par with, or even superior to, corresponding polymer conjugates containing the antineoplastic cisplatin-type structural system. With toxic effects likely to be at a comparatively low level, the ferrocene conjugates of this study offer challenging opportunities for the development of metal-containing, polymer-bound drug systems in cancer research.
References
1. H. Ringsdorf, J. Polym. Sci. Polym. Symp. 51, 135 (1975).
2. Briefly reviewed: E.W. Neuse, Macromol. Symp. 80, 111 (1994).
POLYMER-BASED SYSTEMS FOR SITE SPECIFIC GENE DELIVERY AND TRANSFECTION
D. OUPICKY, K.D. FISHER, Y. STALLWOOD, M. OGRIS, V. MAUTNER, L.W. SEYMOUR
CRC Institute for Cancer Studies, University of Birmingham, B15 2TA, United Kingdom.
Efficient gene delivery and expression holds the key to successful gene therapy for many major diseases, and synthetic polymers play an important role in both non-viral and viral gene delivery systems.
Within the field of non-viral gene delivery we have shown that polyelectrolyte complexes, formed by complexation of plasmid DNA with cationic polymers, can be stabilised by the linkage of multivalent linear copolymers based on N-(2-hydroxypropyl)methacrylamide (HPMA) bearing reactive 4-nitrophenoxy groups. Stabilisation leads to decreased protein binding, greater solubility and improved circulation after i.v. administration to mice. Linkage of targeting agents to the surface of the polymer-coated complexes can lead to selective uptake by receptor-positive target cells, resulting in targeted expression of transgenes.
Adenoviruses can be surface-modified in a similar way, producing ”stealth” viruses. These polymer-coated viruses (pc-viruses) evade antibody recognition and their ability to infect cells via their normal receptors is abolished. Linkage of desired targeting agents to the surface of pc-viruses restores infectivity to receptor-positive cells and leads to efficient transgene expression. Adenoviruses retargeted in this way can infect receptor-positive target cells specifically, and show highly efficient infection of tumour cells in vivo following administration to mice bearing intraperitoneal tumours. This approach should lead to development of viruses capable of infecting specific cells, such as cancer cells, and again should be suitable for systemic delivery.
Non-viral systems for gene delivery are expected to comprise the preferable vectors for use in the long term, exhibiting better safety profiles and ease of scale-up synthesis, while viral vectors, particularly pc-viruses, show the advantage of high efficiency of transgene expression and are likely to be more useful in the short-to-medium term.
This work is supported by the Cancer Research Campaign
DESIGN AND SYNTHESIS OF HIGH EFFICIENCY SYNTHETIC GENE DELIVERY SYSTEMS
D. PUTNAM, C. GENTRY, R. LANGER
Department of Chemical Engineering E25-342, Massachusetts Institute of Technology, Cambridge, MA 02139, U.S.A.
INTRODUCTION: With the advent of modern molecular biology and the potential to not only identify the genetic basis of disease, but to potentially treat diseases at the genetic level, came the promising field of gene therapy. However, while new targets for gene therapy are discovered at an extraordinary pace, the discovery of safe and effective gene delivery vehicles has lagged behind. Currently, there are approximately 400 gene therapy protocols in clinical trials worldwide and the success of these clinical trials is directly influenced by the efficiency of the delivery system.
RESEARCH APPROACH: We have adopted a rational design and synthesis approach to the generation of gene delivery systems. As an initial emphasis we focused on barriers to gene delivery on the cellular level and considered two critical barriers to the transfer of genetic material to the nucleus: DNA condensation and endosomal escape. An additional constraint to our focus was biocompatibility because following gene transfer the cell must be able to support the processes of transcription and translation.
DESIGN and SYNTHESIS: Polymers with high cationic densities (such as polylysine) are well-known to condense DNA into nanostructures via Coulombic interactions. However, high cationic density also leads to cytotoxicity. In addition, imidazole moieties are hypothesized to help mediate endosomal escape. Polymers based on polylysine were synthesized with varying mole percentages of cationic centers interdispersed with imidazole moieties. The synthesis was conducted by polymer analogous reaction of polylysine and 4-imidazoleacetic acid using EDAC/NHS conjugation. Purification was conducted by ultrafiltration. The structure/function balance between cationic density and imidazole content was determined with respect to DNA condensation, cytotoxicity and protein expression.
RESULTS: Polymers were synthesized containing 73.5 mol%, 82.5 mol% and 86.5 mol% imidazole substitutions. Each of the conjugates condensed DNA into structures on the order of 150 nm or below. Using multiple cell lines that are relevant to potential in vivo therapeutic targets (NIH 3T3 fibroblast, HepG2 hepatoblastoma, P388D1 macrophage, and CRL1476 smooth muscle) we have found that with increasing imidazole content, the cytotoxicity of the polymers decreases and luciferase expression from the pCMV-luc increases. At polymer concentrations as high as 0.1 mg/mL the cell viability in vitro is 90% or greater, whereas the viability of cells incubated with polylysine are 20% or lower. Luciferase expression levels are on the order of 9 x 108 light units/mg total protein in multiple cell lines (listed above) and are equal to or better than both Lipofectamine and polyethyleneimine-mediated luciferase expression. These polymers are currently under investigation in vivo for a number of potential disease applications.
PHYSIOLOGICAL CONSIDERATIONS IN DOSAGE FORM DESIGN
P.-Y. YEH, P. L. SMITH
Drug Delivery Department, Pharmaceutical Technologies R&D, SmithKline Beecham Pharmaceuticals, 1250 South Collegeville Road, PO Box 5089, Mail Code UP1230, Collegeville, PA 19426-0989, USA
Oral dosage form design has traditionally focused on the physicochemical aspects of the formulation, such as salt form, excipient, polymorphism, particle size, stability, dissolution rate, etc. These aspects, while very important to consider in formulation development, only account for the means of "presenting" drug molecules in a soluble form to the surface of mucosal membranes. It has long been recognized that absorption across intestinal mucosal membranes is often the rate-limiting step for systemic delivery of therapeutic agents. Our understanding of the factors, which limit transport across mucosal membranes, has developed in part from in vitro methodologies. From in vitro studies, it is now well understood that transepithelial movement of drugs occur via different pathways (e.g., passive diffusion, carrier-mediated, and active secretion). Such understandings are being further expanded with the advances made in pharmacogenomics. Cloning and expression of drug transporters provide tools to drug discovery scientists for optimizing candidate selection. Our increased awareness of these physiological factors should provide the impetus for more efficient candidate selection and physiological based dosage form design in the future.
DESIGN OF THE CONJUGATE OF SUPEROXIDE DISMUTASE (SOD) WITH THE COPOLYMER OF DIVINYL ETHER AND MALEIC ANHYDRIDE AS A ANTI-INFLAMMATORY AGENT
T.HIRANOa, T.KONDOb, T.TODOROKIb, S.OHASHIa
a National Institute of Bioscience and Human Technology, Agency of Industrial Science and Technology, Ministry of International Trade and Industry, Tsukuba-Higashi 1-1, Ibaraki, 305-8566 Japan
b Institute of Clinical Medicine, Tsukuba University, Tsukuba-Tenoudai 1, Ibaraki, 305-8575 Japan
As well known the Human Genome Project (HGP) is going to be the final stage in 2000, and will be completed within these 3 years. The information of the base sequence of human chromosome will be opened and the use of such information will be accelerated as post-genome research. It is considered that most of the human gene, probably about the number of 100,000, will be clarified within these 5years. Each new gene is corresponding to a new protein, which can be either drug candidate itself or the target for drug design. Therefore the number of the protein for drug usage will be drastically increase in the post-genome era, and the necessity to develop appropriate drug delivery system for protein becomes more serious. Drug delivery system for protein is less expensive, and much safer than gene therapy.
We tired to design the polymeric conjugate of superoxide dismutase (SOD) for the use as an anti-inflammatory agent. SOD is known to hydrolyze superoxide anion, which is potential hazard for inflammation, aging, as well as cancer. Conventional Cu,Zn SOD was used clinically for the treatment of rheumatoid, but was not so effective as expected due to the short half life time, less than 5 minutes, in vivo. By conjugation with synthetic polymers the half life of SOD could be increased about 10 times, and the antigenicity of bovine SOD was drastically decreased. The decrease of the enzymatic activity by polymeric conjugation was significantly improved by using a amino protecting agent, 1,2-dimethylmaleic anhydride, which can be quickly removed by lowering the pH of the reaction to 6.0. The copolymer of divinyl ether with maleic anhydride was used for the conjugation of SOD, as it has anti-inflammatory activity itself, and stabilizes the Cu,Zn metal leakage due to the chilating activity.
The anti-inflammatory activity was examined in vivo against animal models. In rat re-expansion model of lung, the attachment of leukocyte on the endotherial surface was drastically inhibited, which results in the inhibition of edema of lung, caused by the damage by oxygen anion radicals. Also the protecting activity of SOD-polymer conjugate was observed in rat liver reperfusion injury model. The obstruction of blood flow as well as leukocyte adhesion in the blood vessels was inhibited by SOD-polymer conjugate, which was shown by real time observation of liver under fluorescent microscope. In both animal models SOD alone showed no significant biological activity. As the liver reperfusion model simulates the process of liver transplanatation, SOD-polymer conjugate can be applied for such purpose to enhance the quality of the transplanted organ. SOD alone has been already applied at the organ transplantation, and the complication after operation was reported to be reduced. Because polymeric conjugate of SOD had contributed more effectively in the reperfusion injury than SOD, it will be useful for clinically.
SL21