Special lectures: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
H. KEUL, A. NEUMANN, B. REINING, H. HÖCKER
Lehrstuhl für Textilchemie und Makromolekulare Chemie der Rheinisch-Westfälischen Technischen Hochschule Aachen, Worringerweg 1, 52056 Aachen, Germany
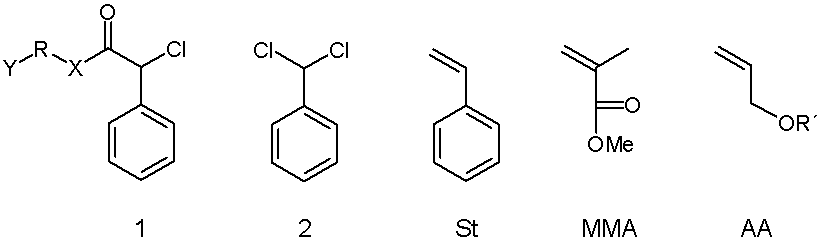
The atom transfer radical polymerization (ATRP) of styrene (St) and methyl methacrylate (MMA) with a macroinitiator 1 (RX = (CH2CH2O)n; Y = OCH3) based on poly(ethylene oxide) leads to block copolymers with a controlled molecular weight and a narrow molecular weight distribution.
Polymerization of St and MMA with a low molecular weight initiator 1 (Y = OH, R = (CH2)n, and X = O or NH) leads to a well defined heterotelechelic product with the endgroups defined by the initiator used.
Polymerization of both St and MMA with benzylidene chloride (2) as initiator leads to a chlorotelechelic product; however, in the polymerization of St a monodirectional chain growth and in the polymerization of MMA a bidirectional chain growth is observed.
The atom transfer radical polymerization of both St and MMA can be terminated by endcapping the chains with an w-functional a-olefin as for example the endcapping agent AA (R´= H or CO-NH-C6H4CH3). Thus, additional functional groups can be introduced as the chain end(s) by a new type of a chain analogous reaction, the atom transfer radical addition.
François SIMAL,* Albert DEMONCEAU, and Alfred F. NOELS
Laboratory of Macromolecular Chemistry and Organic Catalysis
University of Liège, Sart-Tilman (B.6a), B-4000 Liège
The controlled free radical polymerization has in very recent years revitalised the rather mature field of radical olefin polymerization in an unprecedented way, providing access to well-defined polymers and copolymers. Stable free radicals, such as nitroxides, have been introduced for controlling radical polymerization. Recently, the groups of Matyjaszewski, Sawamoto, Jérôme and others have replaced the stable nitroxide free radical with transition metal species to obtain inter alia a variety of copper-, iron-, nickel-, palladium-, or rhodium-mediated controlled free radical polymerization systems, a methodology going by the name of "atom transfer radical polymerization" (ATRP).
Ruthenium has been introduced by Sawamoto, but RuCl2(PPh3)3 (the most widely used ruthenium complex) requires the presence of a Lewis acid activator. We now report on the exceptional efficacy of new catalytic systems based on well-defined and fully characterized RuCl2(p-cymene)(PR3) complexes (1) (p-cymene = 4-isopropyltoluene) for promoting the controlled free radical polymerization of vinyl monomers without activation with co-catalysts. With methyl methacrylate as a model substrate and ethyl 2-bromo-2-methylpropionate as the halogenated initiator, these readily prepared and air stable catalysts compare favourably with the most active ATRP catalysts reported to date in the literature.
On the other hand, [RuCl2(=CHPh)(PCy3)2] (2), the Grubbs ruthenium-carbene complex used commonly for olefin metathesis, has also been shown to be an efficient catalyst for the controlled radical polymerization of vinyl monomers. Under typical experimental conditions, polymerization of methyl methacrylate yielded PMMA with low molecular weight distributions (Mw/Mn ˜ 1.1) and high initiation efficiency (f ˜ 0.9). All the criteria of living polymerization were fulfilled.
In this communication, we will comment on catalyst design and on the polymerization of other vinyl monomers including other methacrylates, acrylates, and 4-substituted styrenes.
(a) Simal, F.; Demonceau, A.; Noels, A.F. 11th International Symposium on Homogeneous Catalysis (ISHC XI), University of St Andrews, Scotland, UK, 12-17 July 1998. (b) Simal, F.; Demonceau, A.; Noels, A.F. Angew. Chem. 1999, 111, 559-562; Angew. Chem. Int. Ed. 1999, 38, 538-540.
F.L. Duivenvoorde, M.A.J. Schellekens, A. Snijder, C.F. van Nostrum, R. van der Linde
Dep. of Polymer Chemistry and Coating Technology, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands.
Methods to synthesize well-defined functionalized polymers are of high importance for fundamental structure-properties studies in coating applications, as well as for the preparation of coating resins or additives having specially designed architectures. We aim at the preparation of diblock copolymers, via living polymerization techniques, for the use as, for example, polymeric dispersants.
Block copolymers are finding wide application in polymer technology, of which the use as compatibilizers is the best known. In coating formulations block copolymers are frequently used as pigment dispersants. The polymers at least contain one part, also called the anchor block, that strongly adheres onto the inorganic pigment surface, and one non-adsorbing part, also called the buoy block, that should mix with the coating matrix on the molecular scale. In principle, the former block can form a flat structure on the surface, while the buoy block will form a pin or tangle protruding from the surface. In this way the copolymers can be seen as ‘compatibilizers’ between the coating and the pigments (or other filler particles), which may result in improved wetting by the resin. Moreover, the adsorbed block copolymers give rise to a steric stabilization of the pigment dispersion, thus preventing flocculation.
In this paper we give an overview of the work carried out in our laboratory on the controlled synthesis and application of two kinds of block copolymers. Depending on the nature of the buoy block, they are designed for the use in combination with polyester and polyolefinic coatings, respectively. One application will be highlighted, i.e. pigment dispersants for powder coatings.
Another part of our research is the synthesis of telechelic linear and branched or star shaped poly(meth)acrylates by means of Atom Transfer Radical Polymerization. The functional groups are introduced by the use of functional initiators and end-capping methods. Such polymers will be used as binder resins, which crosslink at the functional groups.
William J. Brittain
Department of Polymer Science, The University of Akron
Tethered diblock copolymers composed of styrene and (meth)acrylates have been synthesized. The synthetic route involved the following sequence: 1) surface immobilization of 2-methoxy-2-(4-trichlorosilylphenyl)propane onto a silicate substrate, 2) carbocationic polymerization of styrene, and 3) atom-transfer radical polymerization of the (meth)acrylate. The thickness of the polystyrene layer was controlled by the conditions of the carbocationic polymerization (e.g., Lewis acid catalyst, time, and temperature). The samples of tethered diblock polymers exhibited reversible surface changes in response to solvent treatment. For a sample with a 26 nm polystyrene layer and a 9 nm PMMA layer, the advancing water contact angle increased from 75° (characteristic of PMMA) to 99° (characteristic of polystyrene) after treatment with methylcyclohexane; subsequent treatment with CH2Cl2 returned the contact angle to the original value of 75°. We attributed this contact angle change to reversible changes in the chemical composition at the polymer-air interface. Other tethered diblock compositions will be discussed in addition to AFM analysis of surface morphology.
W.JAEGER, U. WENDLER, J. BOHRISCH, A. LIESKE
Fraunhofer - Institut für Angewandte Polymerforschung, D-14513 Teltow, Germany
Water soluble synthetic polyelectrolytes are polymers of continuously growing interest due to their manyfold applications in both industrial processes and daily life. Investigations of the interaction of these polymers with oppositely charged materials require polymers with well defined structure. This includes as an important part ionically charged polymers with regular structure. Macroinitiated polymerization of cationic vinyl monomers leads to triblock copolymers containing an equal number of hydrophilic nonionic and ionically charged monomer units, which were characterized extensively. Using surface active monomers a new type of micellar polymers with unusual properties results. Depending on the structure of the charged block the new polymers are powerful stabilizers in different heterophase polymerization systems. Several of the micellar polymers have remarkable solubilization capacities.
N-oxyl-mediated controlled free radical homo- and copolymerization of suitable aromatic monomers (4-vinylpyridine, vinylbenzylchloride) results in reactive both homopolymers and AB-block copolymers with narrow MWD. The reactive homopolymers can easily be converted to cationic polyelectrolytes with a wide variety of charge strength and of the ratio of hydrophilic and hydrophobic properties of the monomer unit on an equal level of chain length at low polydispersity, serving as models for precise investigations of structure–property relationships. In the same way different polysulfobetaines and polycarboxybetaines are available and used. Corresponding functionalization of the block copolymers leads to amphiphilic cationic block copolymers containing strong hydrophilic and strong hydrophobic blocks of adjustable length and ratio. These block copolymers are efficient stabilizers in the emulsion polymerization of styrene. The resulting monomodal particles are highly charged and of excellent colloidal stability. In a similar reaction pathway block copolomers containing betaine blocks were synthesized for the first time. The micellization of the different block copolymers was investigated in aqueous systems and organic solvents.
BUILDING CHIRAL MOLECULAR ARCHITECTURES USING LIVING TRANSITION-METAL INITIATED POLYMERIZATIONS
BRUCE M. NOVAK
Department of Chemistry, North Carolina State University, Raleigh, NC 27695
Restricting the conformational degrees of freedom within a polymer's backbone can have the effect of extending the chain and endows the material with a number of interesting properties including high modulus and strength, chirality and liquid crystallinity. In order to access materials with these attributes, we have been interested in developing living, chain growth routes into extended-chain, helical polymers. Although both helical, polyisocyanides and polyisocyanates have widely different conformational and rheological properties. This is due to differences in their respective helix inversion barriers and persistence lengths. For various applications, we have been interested in designing systems possessing the persistence lengths of polyisocyanates but with inversion barriers approaching those observed for polyisocyanides. Our conceptual approach to this problem was to replace the carbonyl group in the polyisocyanate backbone with the more sterically demanding imine group of a polyisocyanide. Toward this end, the polymerization of carbodiimides has been investigated using initiators of the type CpTiCl2X, (where X = –NR2, I, or –OCH2CH3, II) (eq 1).

Evidence collected from both the molecular weight / conversion profiles and kinetics indicate that these are living polymerizations that proceed without chain transfer or chain termination side reactions. Polydispersities are generally ≤ 1.1 and molecular weights can be controlled by adjustments in the monomer to initiator ratio. Polymers formed from optically active monomers show large increases and sign changes in their optical rotations, which is indicative distereoselectivity with respect to helical sense. The CV spectra also agree with this analysis. Annealing said polymers at elevated temperatures greatly increasing the magnitude of the optical rotation. This behavior is consistent with the conversion of the chains from helical conformations formed under kinetic control (kinetically controlled conformation, KCC) to conformations now under thermodynamic control (thermodynamically controlled conformation, TCC). The maximum high temperature optical rotation measured for these polymers depends on the temperature (Figure 1). We ascribe this variance to the change in the P/M equilibrium constant at these various temperatures.
S. P. Armes, V. Bütün and N. C. Billingham
School of Chemistry, Physics and Environmental Science, Sussex University, Falmer, Brighton, E. Sussex, BN1 9QJ, UK.
Shell cross-linked ‘knedel’ (SCK) micelles with hydrophilic cores were synthesised using a 2-(dimethylamino)ethyl methacrylate-2-(N-morpholino)ethyl methacrylate [DMAEMA-MEMA] diblock copolymer in aqueous media at elevated temperature.1 The DMAEMA residues in the micelle corona were then cross-linked using 1,2-bis-(2-iodoethoxy)ethane (BIEE). Dynamic light scattering confirmed that the SCK micelle structure was retained. NMR studies showed that the extent of (de)hydration of the MEMA residues in the micelle core could be controlled by varying the solution temperature.
Zwitterionic SCK micelles of 18-25 nm diameter were readily prepared2 from DMAEMA-based block copolymers, again using BIEE as a bifunctional cross-linker in aqueous solution. Depending on the reaction sequence utilised, two classes of shell cross-linked micelles were obtained: Type I micelles, which have anionic cores and cationic coronas, and Type II micelles, which have cationic cores and anionic coronas. Both micelles exhibited isoelectric points in aqueous solution.
Finally, another tertiary amine methacrylate diblock copolymer based on MEMA and 2-(diethylamino)ethyl methacrylate [DEAEMA] was shown3 to form both micelles and reverse micelles in aqueous solution at 20oC. This behaviour, which has never been previously reported for any surfactant or block copolymer, depends on subtle variations in the solution pH and electrolyte concentration.
References
1. V. Bütün, N. C. Billingham, S. P. Armes, J. Am. Chem. Soc., 1998, 120, 12135.
2. V. Bütün, A. B. Lowe, N. C. Billingham, S. P. Armes, J. Am. Chem. Soc., 1999, in press.
3. V. Bütün, N. C. Billingham, S. P. Armes, J. Am. Chem. Soc., 1998, 120, 11818.
Hajime Yasuda
Department of Applied Chemistry, Faculty of Engineering, Hiroshima University,
Higashi-Hiroshima 739-8527, Japan
Living polymerization of methyl methacrylate has been already realized using rare earth metal complexes such as SmMe(C5Me5)2(THF) to give syndiotactic (> 96%) high molecular weight polymers (Mn >1500,000) with very low polydispersity (Mw/Mn < 1.05) in high yield (JACS, 114, 4908, 1992). After expending many efforts, we have succeeded in the preparation of isotactic polymers of methyl methacrylate (isotacticity, > 97%) to give high molecular weight (Mn > 500,000) with low polydispersity (Mw/Mn < 1.13) in high yield (> 96% yield). Characteristics of this type of living polymerization reveal the followings. 1)Stereo complex is formed by mixing the acetone solutions of syndiotactic polymer and isotactic polymer. Resulting stereo complex shows the Tm of 204 °C and annealing of the stereo complex at 200 °C give rise to the formation of the polymer with higher melting point, 230 °C. 2) Block copolymerizations of ethylene with polar monomers such as methyl methacrylate or caprolactone are available. Polymerization of ethylene followed by the addition of excess methyl methacrylate provides block copolymers in high yield 3) Block copolymerization of methyl methacrylate/butyl acrylate/methyl methacrylate gives rubber like elastic polymers. 4) Adhesive materials are obtained by hydrolysis of block copolymer of methyl methacrylate/trimethylsilyl methacrylate.
F. BANDERMANNa, M. FERENZa, R. SUSTMANNb, W. SICKINGb
aInstitut für Technische Chemie, Universität Essen, D-45117 Essen, Germany bInstitut für Organische Chemie, Universität Essen, D-45117 Essen, Germany
The polymerization of methyl methacrylate (MMA) in toluene with Cp2ZrMe2 (1) and Ph3CB(C6F5)4 (2) as initiating system was investigated kinetically. Reaction temperature was 0 oC, the concentration of MMA 1 mol/L, that of the initiators about 1 mmol/L. This system can be regarded as a combination of the systems described by Collins and Soga.
With a ratio (1)/(2) of > 1 monomer, conversions of more than 99 % and syndiotactic-rich poly(methyl metthacrylat) with number-average molecular weights of > 200.000 g mol-1 and dispersion indices of 1.5 - 1.6 were obtained. For ratios £ 1 no polymerization was observed.
We also succeeded in synthesizing the neutral complex Cp2ZrMe[OC(OMe)=CMe2] (3). It was not active in MMA polymerization without further activating compounds. However, in combination with equivalent amounts of (1) and (2) it delivered PMMA with number-average molecular weights of > 100.000 g mol-1 and with dispersion indices of < 1.1. The stereoregularities of the resulting polymers are identical to those observed by using only (1) and (2).
Thus it turned out that both a neutral zirconocene enolate and a zirconocene cation are necessary in the propagation reaction. This result is consistent with the mechanism proposed by Collins et al. A kinetic analysis of time-conversion curves using the PREDICI software package makes such a mechanism probable. However, the mechanism has to be extended by incorporating a first-order termination reaction of the active species. Rate coefficients of all elementary reactions were determined from time-conversion curves.
The kinetic investigations were accompanied by ab initio calculations, which support the assumed kinetic model.
NEW EPOXI INITIATORS FOR THE CONTROLLED SYNTHESIS OF FUNCTIONALIZED POLYISOBUTYLENES
JUDIT E. PUSKAS
Department of Chemical and Biochemical Engineering, The University of Western Ontario, London Ontario, Canada N6A 5B9
It has recently been discovered that substituted epoxides can initiate the living polymerization of isobutylene (IB). Epoxidized a -methylstyrene (MSE) and hexaepoxi squalene (HES) in conjunction with TiCl4 were shown to be effective initiators:


MSE HES
The polymerizations were found to be living, demonstrated by linear first order monomer consumption plots and linearly increasing MWs and narrowing molecular weight distributions MWDs with conversion for both initiators. Controlled initiation with MSE/TiCl4 was proven by triple detection (RI, MALLS and UV) SEC and pyrolysis GC-MS. 1H NMR of representative samples showed the presence of tert-Cl end groups in the PIB. Model experiments with diisobutylene yielded a primary-OH head group as shown below:

HES/TiCl4 was shown to produce branched polyisobutylenes (PIBs) with Mn > 400,000 and MWD <1.2. Integration of the gem-dimethyl 1H NMR signal of the tert-Cl end groups yielded 5.1 arms per molecule. Diphenyl ethylene (DPE) end capping resulted in the disappearance of the gem-dimethyl signal, and integration of the DPE-signals yielded 5.2 arms per molecule.
The kinetics and mechanism of the initiation and polymerization were also studied by in-situ fiberoptic mid-IR spectroscopy in real-time.
A. MADL, S. SPANGE
Department of Polymer Chemistry, Institute of Chemistry, University of Technology Chemnitz, Strasse der Nationen 62, D-09111 Chemnitz, Germany
Vinylformamide (VFA) is a suitable precursor monomer for the synthesis of poly(vinylamine) (PVAm), a potential polyelectrolyte. The cationic polymerization of VFA is initiated by means of iodine, bromine, trifluoromethane sulfonic acid (HOTf), and trimethylsiliyl triflate (TMST) in toluene at different reaction temperatures. Chain structures, head group functionality, and MWD of the oligo(vinylformamide) (OVFA) are investigated by 1H NMR spectroscopy, MALDI-TOF-MS, GPC, and quantitative elemental analyses.
OVFAs with narrow MWD are obtained in moderate yield (5 – 50 %) in the temperature range from 253 K to 313 K. Reaction temperature below 253 K is not suitable for the cationic polymerization of VFA because no oligomeric products are yielded. The yields, properties, and chemical constitutions of the OVFAs, such as the average molecular weights and the chain structures, are strongly determined by the reaction temperature used. With increasing reaction temperature the head group functionality decreases and the yield of OVFA increases.
The acid or base induced hydrolysis of the OVFAs leads to oligo(vinylamine) (OVAm). The structure of the OVAms, based on different precursor OVFAs, is investigated by 1H NMR and 13C NMR spectroscopy and MALDI-TOF-MS. The high reactivity of the amino groups of the oligomer is the key for chemical modifications. Applications of the OVAms as polyelectrolyte compound of hybrid materials are presented.
MOLECULAR RECOGNITION IN FREE-RADICAL TEMPLATE POLYMERIZATION
JOANNA SZUMILEWICZ
Department of Physical Chemistry of Polymers, Technical University of Łódź
ul Żeromskiego 116, 90-543 Łódź, Poland
Free-radical template polymerization leads to formation of interpolymer complexes. The complexes are products of interactions of template macromolecules and macromolecules of daughter polymer.
In our previous work we have found existence of a molecular recognition effect with regard to chain length during polycomplex formation when mixing polymer solutions in a common solvent. In a selected set of circumstances macromolecules of one of polymer components can be recognized in such a manner that only the longest chains present are bound in form of the complex.
In the present work we found a similar phenomenon to occur in a template polymerization with regard to template polymer. This is a new aspect of template polymerization reaction. The system studied was template polymerization of methacrylic acid on poly(N,N’-dimethylanimoethyl methacrylate) (PDMAE). The polymerization, carried out in tetrahydrofuran or in ethanol, was initiated by AIBN. A series of copolymerizations of methacrylic acid and methyl methacrylate on PDMAE was performed under similar conditions.
Changes in molecular weight distribution (MWD) of PDMAE template in course of copolymerization were studied by means of gel permeation chromatography method (GPC). A method had been worked out that permitted simultaneous determination of MWD of the complexed fraction of the template as well as of the extent of polymerization of comonomers and the composition of the polycomplex formed. It was found that the recognition effect with regard to chain length was observed in the case of copolymerization as well in homopolymerization of methacrylic acid. The results obtained are discussed as related to the composition of the polycomplexes formed and to the template – daughter polymer interaction. The effect of solvent used was also taken into account.
New surfmers and inisurfs for polymer particles with new morphology
Hans-Juergen P. Adler°, Carsten Puschke°, Andrej Pich°, Karsten Jahny°, Stanislav Voronov°°, Meifang Zhu°°°
°Institute for Macromolecular Chemistry and Textile Chemistry
University for Technology of Dresden, Germany
°°State University of Lviv, Ukraine
°°°China Textile University of Shanghai, China
Polymeric nano-particles with well-defined colloidal and surface characteristics have obtained increasing attention for different purposes, e.g. in coatings, adhesives, in biomedical fields and as fillers in blends.
The morphology of particles is variable in a broad range by two-stage emulsion polymerization to core-shell-particles. By this way special reactive groups are introducable in the shell for different applications.
The core-shell morphology can be built up in a batch or semi batch procedure or by seed polymerization. In the second case the particle morphology is determined mainly by the polarity and the compatibility of the shell and core polymer.
We synthesized core-shell particles by the direct grafting of a shell monomer onto the surface of seed particles which contained labile peroxy groups. The synthesis of peroxy- functionalized core particles was carried out using two methods:
The amounts of peroxide groups as well as the thickness and structure of the shell were varied and characterized by different methods.
The kinetic of thermal decomposition of PPE was studied successfully by DCS and Raman Spectroscopy. The ESR- spectroscopy showed only qualitative results due to the weakness of signal intensity. We studied the kinetic of emulsion polymerization of different monomers in presence of PPE as “Inisurf” for calculation the activation energy and te rate of reaction.
By these ways peroxide contained particles are available which are reactive fillers in a surrounding matrix and also to form special onion- like core-shell- morphologies with different shells. Freeze dried dispersions could be used as a reactive powder mixed with other polymer materials and form blends with covalent bounds between the filler and matrix.
Mixed peroxide containing dispersions with other dispersions, like polybutadiene dispersions, formed crosslinked polymer latex films with improved properties, like elasticity and /or hardness depending on the content of reactive dispersions, temperature and time of heat treatment.
Another way to core-shell-structures was in using polyurethane macromonomers as “Surfmers”, which we could copolymerize with styrene or acrylates.
As the result a new kind of polyurethane dispersions with a polyurethane shell and polyurethane- acrylate copolymer core we could obtain. These particles are interesting for waterborne coatings
SYNTHESIS AND CHARACTERIZATION OF ACRYLATE FUNCTIONALIZED UNSATURATED POLYESTERS
Jasna Djonlagic, Alisa Zlatanic, Branko Dunjic
Faculty of Technology and Metallurgy, University of Belgrade, Karnegijeva 4, Belgrade 11000, YUGOSLAVIA
The polycondensation reaction of potassium dicarboxylate with 1,4-dibromobutane, which has been successfully applied to the synthesis of stereoregular unsaturated polyesters and copolyesters1,2, was extended to the preparation of acrylate-terminated copolyesters3. Three series of acrylate functionalized random copolyesters poly(tetramethylene maleate-co-tetramethylene phthalate), poly(tetramethylene fumarate-co-tetramethylene phthalate) and poly(tetramethylene phthalate-co- tetramethylene succinate) were synthesized. All these polycondensation reactions take place in heterogeneous reaction systems, in which the insoluble potassium salts of the acids form the solid phases and 1,4-dibromobutane is soluble in 1-methyl-2-pyrrolidone (NMP). The monofunctional potassium salt of acrylic acid was used for the introduction of double bonds at the end of the chains and for the regulation of the molecular weight of the polyesters.
The telechelic copolyesters were characterized by 1H and 13C NMR spectroscopy, in almost all cases providing an acrylate functionality of two. The average molecular weights of the synthesized polyesters were determined by vapor phase osmometry (VPO), gel permeation chromatography (GPC) and by analysis of 1H NMR spectra. The results suggest that the applied procedure of polyester synthesis can be used for the introduction of acrylate groups at the chain ends, but for fine regulation of the molecular weight it would be necessary to take into account the solubility of the potassium salt of acrylic acid.
The introduction of acrylate groups at the end of the polyesters chains diminishes the number of dangling chains and allows the formation of a more regular network structure by the crosslinking copolymerization with styrene. Also, the influence of these terminal acrylate groups and cis-trans configuration of the double bonds along the polyester chain was evaluated by the chemorheology of the curing behavior and by the viscoelastic properties of these cured unsaturated polyesters.
References
1. N. Lacoudre, A. Leborgne, M. Sepulchre, N. Spassky, J. Djonlagic, M.S. Jacovic, Makromol. Chem., 187 (1986) 341.
2. J. Djonlagic, M.O. Sepulchre, M. Sepulchre, N. Spassky, B. Dunjic, M.S. Jacovic, Makromol. Chem., 191 (1990) 1529.
3. A. Zlatanic , J. Djonlagic, Macromol. Chem. Phys., 198 (1997)1775.