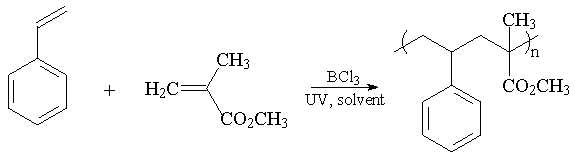
Posters: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61
D.C. SHERRINGTON AND K. TASKINEN
University of Strathclyde, Department of Pure & Applied Chemistry, Thomas Graham Building, 295 Cathedral Street, Glasgow, G1 1XL, Scotland, UK
Methyl methacrylate is an example of a good acceptor and its electron deficiency can be further reduced using Lewis acids, i.e. BCl3 forms a complex with methyl methacrylate. This is believed to be crucial for formation of a regular 1:1 structure in styrene and methyl methacrylate copolymers. Copolymers of styrene and methyl methacrylate have been produced using BCl3 as a Lewis acid. Depending on the ratio of the BCl3 compared to methyl methacrylate different yields and compositions in the region of 50:50 have been obtained. Using the spectroscopic criteria in the literature all the soluble copolymers have been shown to have an alternating structure. The UV-initiated copolymerisations have been carried out in toluene, dichloromethane and hexane, at different temperatures and using different reaction times and with different ratios of BCl3 compared to methyl methacrylate under. After optimisation of the reaction conditions, the best yield, 57 %, was achieved using 12.5 % BCl3 at -25 °C in dichloromethane.
Fig.1: Alternating copolymerisation of styrene and methyl methacrylate
Our results show there is a window of BCl3 concentrations that allows the synthesis of alternating copolymers in useful preparative amounts. The yields of alternating copolymers are at best about 50 % which are very much higher than any data reported in the literature. Most products show a bimodal molecular weight distribution. Of the Lewis acids examined only BCl3 gave practical yields of alternating 1:1 copolymers of styrene and methyl methacrylate.
L. CORVAZIERa, Y. ZHAOb
a Chemistry Department, Sheffield University, Dainton Building, Brookhill S3 7HF Sheffield, ENGLAND
b Chemistry Department, Sherbrooke University, Sherbrooke, J1K 2R1, P.Q., CANADA
A diacrylate monomer (fig. 1) containing a core of azobenzene group was synthesized. Radical polymerizations of the monomer dissolved in a nematic liquid crystal, E7, resulted in liquid crystals stabilized by an azobenzene-containing polymer network. When their thin films were irradiated by a linearly polarized UV light at room temperature, as a result of the alignment of azobenzene groups on the network, a macroscopic orientation of E7 molecules was induced perpendicular to the UV polarization direction. Infrared dichroism was used to measure the liquid crystal orientation in irradiated films. It was found that the achievable orientation, observed after minutes of irradiation, increases with increasing the polymer network density, and that a significant degree of the induced orientation remains stable after turning off the irradiation. This approach represents a new means to control the average liquid crystal orientation in materials like polymer-stabilized liquid crystals.
I.PROKOPOVÁa, M.MAREKb, J.DVOŘÁČEKb, E.VLČKOVÁa
aDepartment of Polymers
bDepartment of Food Preservation and Meat Technology
Prague Institute of Chemical Technology, Technická 5, 166 28 Prague 6
Czech Republic
Aliphatic polyamides have structural units similar to natural polymers – proteins. However, synthetic homopolyamides are from the reason of their over-molecular structure practically eliminated from the group of biodegradable materials. The copolymerization of 6-hexanelactame with L-lysine or L-alanine yields copolymers which showed in the test of aerobic biodegradation (by means of microbial strains Acinetobacter and Klebsiella) higher sensitivity to biodegradation in comparison with polyamide 6. The effect of the reduced crystalline content in polyamides upon a decrease in resistance to a biological degradation of these materials has been significantly proved also in the case of copolymers of 6-hexanelactam with 12-dodecanelactam. Biodegradable copolyamides can find (likewise copolyesters) interesting applications, e.g. in combination with materials from recyclable natural resources. For such applications copolyesters on the base of polyethyleneterephthalate modified with L-lactic acid also can be appropriate. Copolyesters having 20-40 mol% of incorporated structural units of L-lactic acid have been prepared.by the reaction of L-lactic acid water solutions with polyethyleneterephtalate in a two step process at the temperature range from 220 to 2500C. A successive biological decomposition of these copolymers has been proved by means of microorganisms present in compost soil.
Copolyamides and copolyesters with enhanced sensitivity to bio-degradation have good assumptions for the application in the branch of surface hydrophobization of packaging materials on the base of thermal modified starch. It has been demonstrated that the hydrophobizing layer of the copolymer did not prevent a biological degradation of polysaccharides and by itself has been decomposed at a sufficient rate. The biodegradability of these combined packages has been measured on the base of CO2 production by their exposition in compost soil.
MAYUMI HAYASHI, AKIRA HIRAO
Department of Polymer Chemistry, Tokyo Institute of Technology, 2-12-1, Ohokayama, Meguro-ku, Tokyo 152-8552, JAPAN
The synthesis of well-defined polystyrenes functionalized with one, two, three, and four chloromethylphenyl groups at the chain ends or in the chains by means of anionic living polymerization in conjunction with functional group transformation are described.1) The synthetic method involves the introduction of the anion-stable methoxymethylphenyl groups at the chain ends and in the chains and subsequent transformation reactions into highly reactive chloromethylphenyl groups with BCl3 in quantitative yields.
Furthermore, a new iterative approach using 3-(t-butyldimethylsilyoxy)-1-propyllithium is proposed for the synthesis of well-defined polystyrenes having two, four, and six methoxymethylphenyl and six chloromethylphenyl termini. It involves repeated chemical transformations at the chain end from t-butyldimethylsilyloxybutyl, bromobutyl, methoxymethylphenyl to chloromethylphenyl group. At each stage in the iteration, the number of chloromethylphenyl group doubles.
The resulting polymers all, after HPLC fractionation, possessed well-controlled molecular weights and narrow molecular weight distributions. Their functionalities were nearly quantitative in all cases.
1) Hayashi, M.; Hirao, A. Macromolecules 1999, 32, 0000.
M. KOBAYASHI and S. NAKAHAMA
Department of Polymer Chemistry, Tokyo Institute of Technology, 2-12-1 Ohokayama Meguro-ku Tokyo, JAPAN
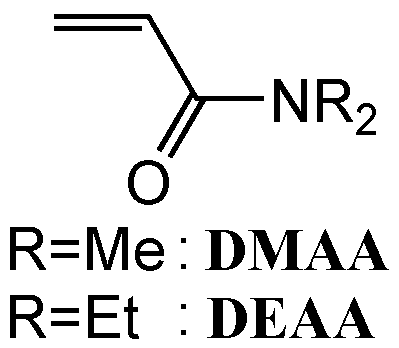
Anionic polymerizations of N,N-dimethyl (DMAA) and N,N-diethylacrylamide (DEAA) were carried out under a high vacuum condition with various in itiators in the absence and presence of Lewis acid in THF for 60 min. All the polymers were obtained in quantitative yields. DMAA and DEAA were polymerized with sec-BuLi/diphenylethylene (DPE) to give the isotactic rich polymers having very broad molecular weight distribution(MWD), while the polymers generated in the presence of weak Lewis acid such as triethylborane (BEt3) or diethylzinc (ZnEt2) had narrow MWDs and highly syndiotactic configuration. In the case of diphenylmethyl potassium (Ph2CHK) modified with Lewis acid, the polymers with very narrow MWDs were produced even at an ambient temperature. Especially, in the case of K+/ZnEt2 system, the polymerization proceed with living nature in THF even at 0 ° C, which is verified by the results of postpolymerization and block copolymerization. We suppose that the Lewis acid coordinates with the propagating amide enolate anion to lower its activity reducing the propagation rate. Interestingly, the combination of diphenylmethyl Grignard reagent and ZnEt2 produced the poly(DEAA) having significantly high degree of isotacticity (mm=91%) and was narrow MWD (Mw/Mn=1.86).
Table 1. A nionic Polymerization of N,N-Dialkylacrylamide in THF for 60 min.
Additive |
Temp. |
Mn X 10-3 |
Tacticity, % |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Initiator | (Add./Ini.) |
Monomer |
° C |
Calc. |
Obs. |
Mw/Mn |
mm |
mr |
rr |
| sec-BuLi/DPE | DMAA | -78 |
9.4 |
11 | 2.58 |
63 |
12 |
25 |
|
| sec-BuLi/DPE | DEAA | -78 |
9.9 |
11 | 2.83 |
60 |
12 |
28 |
|
| sec-BuLi/DPE | BEt3 (2) | DMAA | 0 |
16 |
16 | 1.13 |
0 |
15 |
85 |
| sec-BuLi/DPE | ZnEt2 (18) | DEAA | -78 |
9.6 |
11 | 1.12 |
0 |
12 |
88 |
| Ph2CHK | ZnEt2 (18) | DMAA | 0 |
11 |
9.8 | 1.07 |
2 |
27 |
71 |
| Ph2CHK | ZnEt2 (17) | DEAA | 0 |
41 |
40 | 1.05 |
3 |
89 |
8 |
| Ph2CHK | BEt3 (2) | DMAA | 30 |
14 |
12 | 1.05 |
3 |
20 |
77 |
| Ph2CHK | BEt3 (2) | DEAA | 30 |
9.8 |
10 | 1.21 |
7 |
81 |
12 |
| Ph2CHMgBr | DEAA | -78 |
7.2 |
12 | 4.11 |
86 |
14 |
0 |
|
| Ph2CHMgBr | ZnEt2 (2) | DEAA | -78 |
5.9 |
7.4 | 1.86 |
91 |
9 |
0 |
Yield~100%.
TOYOJI KAKUCHI, NAOYA SUGIMOTO
Division of Biosience, Graduate School of Environmental Earth Science, Hokkaido University, SAPPORO 060-0810, Japan
We have demonstrated the synthetic method for the well-defined end-functionalized polymers with saccharide residues by means of controlled radical polymerization. In this study, the TEMPO-based initiators having glucose, maltose and maltohexose residues, 1, 2, and 3, were synthesized and used as the initiator to produce well-defined end-glycosidated poly(styrene)s. Bulk polymerization of styrene initiated by initiators 1 - 3 was carried out at 120 °C, in which the molar ratio of styrene and initiator ([styrene]/[initiator]) was 600 (Table 1). The molecular weight of the polymers was found to increase in an approximately linear fashion with conversions, while low polydispersities (Mw/Mn) were maintained throughout the polymerization.
Table 1. Bulk polymerization of styrene using initiators 1 - 3 a)
initiator |
time (h) |
yield (%) |
Mn b) |
Mw/Mn b) |
|---|---|---|---|---|
1 |
3 |
21 |
11,400 |
1.12 |
9 |
48 |
22,000 |
1.13 |
|
15 |
59 |
25,200 |
1.13 |
|
2 |
3 |
29 |
18,000 |
1.12 |
9 |
56 |
28,200 |
1.11 |
|
15 |
63 |
31,200 |
1.11 |
|
3 |
3 |
24 |
20,900 |
1.21 |
9 |
46 |
30,400 |
1.15 |
|
15 |
65 |
38,800 |
1.18 |
a) Temp., 120 °C; [styrene]/[initiator], 600.
b) Determined by GPC using a polystyrene standard.
L.M.FRAGA*, V.RUIZ SANTAQUITERIA*
M.C.CARREÑO**, G.FERNÁNDEZ**, C.LAFUENTE**
* Repsol S.A. D. Gral.Tecnología. c/Embajadores nº183, 28045-Madrid
** Dpto. de Química Orgánica, Univ. Autónoma, Cantoblanco 28049-Madrid
Although anionic synthesis of functionalized polymers has been efficiently achieved by functionalized alkyl Lithium initiators, no systematic studies of initiators reactivity have appeared up to date. We report herein the results of the anionic polymerization of styrene, chosen as a model monomer, with different functionalized initiators. Precursors giving rise to the initiators belong to the following groups of organic compounds :
1.- Carboxylic acids and derivatives (oxazolines and amides) precursors of enolate anions.
2.-Sulphur derivatives (methyl thioethers, sulphoxides and sulphones) precursors of I -Li sulphur functionalized carbanions.
3.-Aromatic methoxy and/or amino substituted derivatives precursors of ortho lithiated species.
4.-Phenyl substituted styrene and 1,2-diphenyl ethylene derivatives precursors of I -Li-b-phenylethyl carbanions.
This study evidenced that I -Li anions (group 1) are not able to initiate the polymerization. This lack of reactivity can be attributed to the stabilised structure of the initiators. I -Lithium nitromethane (LiCH2NO2), an enolate analogue, neither produced polymerization.
The results obtained with precursors included in group 4 corroborate the above observations. Addition of n-BuLi to a substituted styrene (p-nitro and o,p-dimethoxyphenyl substituted) or 1,1-di-(p-N,N-dimethylamino)phenyl ethylene yielded a stabilised benzylic anion unable to polymerize styrene.
The behaviour of sulphur containing precursors (group 2) is also dependent on the relative stability of the corresponding anions. Thus I -Li methyl-p-tolyl sulphoxide and the analogue sulphone, did not initiate polymerization whereas I -Li methyl thioether (LiCH2S-p-Tol) gave polystyrene .
The use of 2-Li-1,4-dimethoxybenzene (group 3) did not initiate the reaction, but the more reactive 2,5-dilithium analogue (generated by treatment of 1,4-dimethoxy benzene with 2 eq of n-BuLi) gave polystyrene.
References
G. DAVIDa, V. ALUPEIa, B. C. SIMIONESCUa, C. I. SIMIONESCUb
aDepartment of Macromolecules, ”Gh. Asachi” Technical University, 22, Carol Alley, 6600 Jassy, Romania
b”P. Poni” Institute of Macromolecular Chemistry, 41A, Gr. Ghica Voda Alley, 6600 Jassy, Romania
2-Alkyl-2-oxazolines are known as monomers able to undergo ring-opening polymerization of a living nature under appropriate reaction conditions, thus providing opportunities to control the molecular design of the macromolecular products. End-capping of the living oxazolinium species with various nucleophiles was extensively applied as an efficient approach for the synthesis of macromonomers and telechelics.
Here, 2-methyl-2-oxazoline and benzyl bromide or 1,4-dibromo-2-butene were used as a monomer and initators, respectively, for the obtention of living propagating species, which by reaction with nucleophiles such as maleic acid or cinnamic acid gave the corresponding poly(2-oxazoline) macromonomers or unsaturated polyesters. The coupling reactions were carried out in the presence of two different scavengers, namely: a low molecular weight one, i. e. triethylamine, or a macromolecular one, i.e. poly(4-vinylpyridine-co-divinylbenzene).
Direct termination of living polymer species with another coupling reagent, 1,1-diphenylethylene, was applied in the synthesis of a macromolecular transfer agent.
The functionalized poly(alkyl oxazoline)s were used to synthesize amphiphilic block and graft copolymers or polymer networks.
JENS HØG TRUELSENa, JØRGEN KOPSa, WALTHER BATSBERGb
a Department of Chemical Engineering, Technical University of Denmark, DK-2800 Lyngby, Denmark
b Condensed Matter Physics and Chemistry Department, Risoe National Laboratory, DK-4000 Roskilde, Denmark
A triblock copolymer with well-defined structure prepared by living carbocationic polymerization of isobutylene followed by addition of a smaller amount of p-methylstyrene had n = 61,300 with short endblocks with n = 2,400.
The P(p-MeS)-b-PIB-b-P(p-MeS) was brominated with elemental Br2 in hexane to obtain a product in which 43% of methyl groups was monobrominated with formation of no other brominated groups. The bromomethyl group on the phenylring has previously been found to be a suitable initiating group for ATRP of styrene. The obtained macroinitiator with a degree of polymerization of well over 1000 had end blocks with around 20 repeat units of which on the average 8-9 were bromomethylated.
The P(p-BrMeS-co-p-MeS)-b-PIB-b-P(p-BrMeS-co-p-MeS) macroinitiator was subjected to ATRP of styrene in bulk, under various conditions with CuBr as the transition metal component. 1,1,4,7,10,10-Hexamethyltriethylenetetraamine (HMTETA) was used as the complexing ligand in order to obtain a homogeneous polymerizing system, however, this resulted in formation of a crosslinked product. Due to the low redox potential of the copper(I)-complex with this ligand the polymerization rate is fast with generation of many radicals which also lead to cross-linking. The use of bipyridine as ligand caused the polymerization to proceed in a controlled manner when the conditions were optimized with a gradual increase in Mn over 4 hours to 245,000 while the grafted polymer remained soluble. The polydispersity remained below 2. Simultaneously, however, thermopolymerization occurred although in a controlled manner with formation of homopolystyrene with a Mn increasing to around 10,000 and a polydispersity of 1.1. p-Acetoxystyrene was also polymerized by ATRP in a controlled manner with this macroinitiator although in xylene solution due to the limited solubility of the macroinitiator in this monomer.
J. LUSTON a, J. KRONEKa, F. BÖHMEb, H. KOMBERb
aPolymer Institute, Slovak Academy of Sciences, Dúbravská cesta 9, 84236 Bratislava, Slovakia, e-mail: upollust@savba.sk
bInstitut für Polymerforschung Dresden e.V., Hohe Strasse 6, D-01069 Dresden, Germany
2-Oxazolines represent very interesting group of compounds with a broad utilization in organic synthesis [1] and polymer chemistry [2]. In 60’s, the cationic ring-opening polymerization of 2-oxazolines has been discovered. On the other hand, 2-oxazolines provide also thermally initiated addition reactions with a wide range of reagents, for example with carboxylic acids, phenols, thiols and amines [3]. These reactions provide a base for the preparation of polymers by polyaddition reactions, either by a reaction of bis-(2-oxazoline)s with difunctional reagents or by the construction of ABx monomers containing an 2-oxazoline unit and another functional group. Such monomers would be stable at room temperature and polymerize at elevated temperature.
Reaction between 2-oxazolines and phenols results in ring-opening and formation of ether-amide structure. When bis(2-oxazoline)s and bisphenols are used for the reaction, products of polyaddition reaction are formed [4].
We prepared different compounds of the AB type containing 2-oxazoline unit and a phenol moiety, and used them for polyaddition reactions. Thermally initiated reactions of these compounds lead to the formation of the poly(ether-amide)s. Polymerizations were performed in melt and in a solution. Effect of the monomer structure on the course of polymerization was demonstrated. Presented polymerizations are new type of AB polymerization of functionalized 2-oxazolines.
REFERENCES:
1. Gant T.G., Meyers A.I., Tetrahedron, 50, 2297 (1994)
2. Kobayashi S., Prog. Polym. Sci., 15, 751(1990)
3. Kronek J., Lustoň J., Bö hme F., Chem. Listy, 92, 475 (1998)
4. Jaeger A., Ger. Pat. 1,050,540(1959)
Peter Nesvadba, Marie-Odile ZINK, Andreas KRAMER
Ciba Specialty Chemicals Inc.
CH-4002 Basle
E-mail: peter.nesvadba@cibasc.com
The reaction of the easily available nitrone (I)[1] with AIBN affords the crystalline alkoxyamine (II):
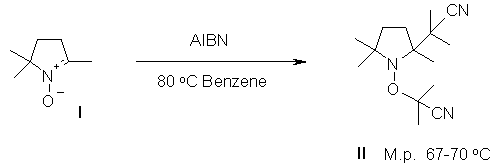
Likewise, the readily accessible 2,2,4-trimethyl-4-nitroso-pentane (III)[2] gives (IV) when reacted with ACCN:
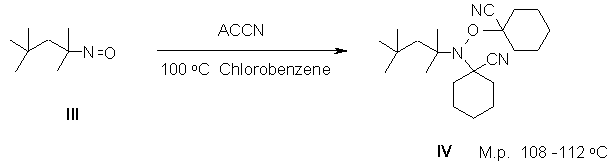
Both alkoxyamines are highly active mediators for the pseudoliving free radical polymerization of styrene. In bulk, for example, (II) affords 60% of polystyrene (1.5mol% II, Mn=3400, PD=1.3) after 3hours at 110 oC. Similarly, (IV) gave 46% of polystyrene (1.5mol% IV, Mn=2700, PD=1.3).
n-Butylacrylate could also be polymerized, however, the degree of control was lower.
References:
[1] Turner, M. J.; Luckenbach, L. A.; Turner, E. L. Synthetic Communications 1986, 16(11), 1377.
[2] Organic Syntheses 1986, 65, 166.
H. JANECZEK, I. BOSEK
Centre of Polymer Chemistry, Polish Academy of Sciences, 34 Maria Curie Sk³odowska St., POB 20, 41-819 Zabrze, Poland
Polymerization reaction of methyl methacrylate (MMA) induced with supramolecular complex of potassium K+,crown,K- was studied [1-3]. It was found that the supramolecular complex of potassium is capable of transferring of two electrons towards to the MMA molecule to form methyl methacrylate carboanion as evidenced by GC-MS, 39K, 13C and 13C DEPT NMR spectroscopy:
MMA
Me+,crown,Me- ¾ ¾ ¾ ¾ ¾ ¾ ¾ ® Me+ + MMA-carbanion
transfer of 2 e-
(where Me is alkali metal e.g. potassium or sodium, crown- is macrocyclic complexant: 18-crown-6 and MMA is methyl methacrylate).
The carboanion of methyl methacrylate is capable of transferring his charge to neutral molecules, thus inducing polymerization of MMA:
MMA-carbanion + MMA ¾ ¾ ® PMMA
The copolymerization of living MMA with e -caprolactone and d -valerolactone is also demonstrated. The structure and properties of obtained polymers were analyzed by 1H, 13C NMR spectroscopy and DSC.
This work was supported by KBN Project No. 3T09A 11513.
References:
1. Z. Jedliñski, Accounts of Chemical Research, 1998, 31, 55.
2. J. L. Dye in Progress in Macrocyclic Chemistry; Izatt R. M, Christiansen I. Eds. Wiley, New York, 1979, vol. 1, p. 63-114; R. P. Edwards, Adv. Inorg. Chem. Radiochem. 1982, 25, 135.
3. Z. Jedliñski, H. Janeczek, I. Bosek, J. Org. Chem., in press; Z. Jedliñski, H. Janeczek, I. Bosek, Macromolecules, in press.
Vinyl polymers with organotin end-groups. Synthesis and modelling
G. Litvinenkoa, A. Arest-Yakubovicha, B. Nakhmanovicha, A.H.E.Müllerb
aKarpov Institute of Physical Chemistry, Vorontsovo pole 10, Moscow 103064, Russia
bInstitute of Physical Chemistry, J. Gutenberg University, Welderweg 15, D-55099, Mainz, Germany
It was observed recently that organotin terminal groups markedly improve dynamic properties of synthetic rubbers1. From this point of view, the use of initiators containing organotin moiety combined with an alkali metal looks very attractive. The kinetics, molecular weight parameters, and the structure of butadiene, styrene, butadiene-co-styrene, and t-butyl methacrylate polymers obtained with new (n-Bu)3SnLi (TBTL) and (n-Bu)3SnNa (TBTS) initiators have been studied.
According to the data of elemental analysis, the butadiene polymers and styrene-butadiene copolymers formed with both TBTL and TBTS contain, on the average, one tin atom per macromolecule. This practically important finding confirms that the initiation proceeds directly via tin-containing initiator rather than via products of its decomposition R3SnMt D R2Sn + RMt as is usually assumed in the chemistry of organotin-alkali compounds2, and, therefore, verifies the usefulness of tin-alkali initiators for improving polymer properties.
Some specific features of polymerization kinetics with tin-containing initiators were observed. In particular, in the presence of Bu3SnNa the initiation rate decreases with increasing medium polarity and an induction period appears upon THF addition; the duration of the induction period is ca. 8 - 11 h in pure THF at 30 °C. The new kinetic scheme was suggested which explains the induction period by the degenerative chain transfer to the initiator precursor. The kinetic scheme proposed enabled us to quantitatively describe the experimental results and was confirmed by model experiments.
1 R.P.Quirk, S.H.Jung, Rubber Chem. Tecnol.,1996, 69, 444.
2 A.G.Davies. Tin. In: "Comprehensive Organometallic Chemistry II" E.W.Abel, F.G.A.Stone, G.Wilkinson, Eds, 1995, 2, 283.
M. Janata, L. Lochmann, J. Brus, J. Brožová, Z. Pientka
Department of Controlled Polymerization, Institute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, Heyrovsky Sq. 2, CZ-162 06 Prague 6, Czech Republic
Nonpolar block copolymers can be multisite-grafted in one block using a method consisting of two steps: (i) selective metalation of the starting copolymer by the complex of sec-butyllithium and N,N,N´,N´-tetramethylethylenediamine in an aliphatic hydrocarbon medium and (ii) in situ grafting of the multi-metalated intermediate with an appropriate nonpolar monomer. Using this method, commercially available Shellvis-type block copolymers composed of polystyrene and hydrogenated diene blocks were selectively grafted with 4-methylstyrene (4-MS) only in the more acidic polystyrene block. Benzene was present in the grafting step in order to prevent transmetalation of methyl group in poly(4-MS) grafts. The graft copolymer prepared in this way was then again selectively grafted using a similar two-step method including : (i) selective metalation of methyl groups in poly(4-MS) grafts by Superbase, made from 2-ethylhexyllithium and potassium tert-pentoxide in benzene and (ii) in situ grafting of the multi-metalated graft copolymer with styrene. In this way, multi-grafted copolymers with polystyrene grafts on poly(4-methylstyrene) grafts were formed. Transmetalation of the metalated intermediate cannot occur under these conditions.
The number of grafts and their length can be varied in a wide range and thus diverse polymer structures are accessible. Multi-grafted copolymers with polystyrene and poly(4-MS) grafts can be selectively functionalized with various functional groups, such as COOH and Si(CH3)3 , using the procedures described above.
All the synthesized grafted and functionalized copolymers were characterized by 1H, 13C and 29Si NMR and by SEC.
Membranes were prepared from the copolymers in order to investigate their transport properties in separations of gas mixtures.
TAMÁS FÓNAGY, BÉLA IVÁN, MÁRTA SZESZTAY
Institute of Chemistry, Chemical Research Center of the Hungarian Academy of Sciences, H-1525 Budapest, Pusztaszeri u. 59-67, P.O.Box 17, Hungary
Poly(isobutylene-g-styrene) (PIB-g-PSt) was synthesized by quasiliving atom transfer radical polymerization (ATRP) of styrene. The ATRP process was induced by an industrially produced poly(isobutylene-co-p-methylstyrene-co-p-bromomethylstyrene) macroinitiator (PIB) in the presence of CuBr and 2,2’-bipyridine (Scheme). Successful graft polymerizations were per-formed both in bulk and in xylene solution. The resulting polymers were characterized by 1H NMR and UV-visible spectroscopies, gel permeation chromatography (GPC) and differencial scanning calori-metry (DSC). It was found that ATRP of styrene with the PIB macroinitiator led to homopolymer free PIB-g-PSt graft copolymers with different compositions. Phase separated morphology in these graft copolymers was found by DSC. Stress-strain analyses indicate high elongation and tensile strength for PIB-g-PSt grafts with certain composition. This new grafting process and the resulting thermoplastic elastomer (TPE) is candidate for several useful applications.
I.POLJANŠEK, UA. ŠEBENIK
Faculty of Chemistry and Chemical Technology, University of Ljubljana, Aškerčeva 5, 1000 Ljubljana, Slovenia
In this study controlled two step block copolymerization of styrene and methyl methacrylate with tetraphenyl biphosphine (Ph4P2) under UV irradiation took place in bulk and in THF solution. In the first step of polymerization PS - and PMMA - macroinitiators were synthesized. The polymers from the first step of polymerization with active phosphine groups on both ends of the polymer chain can be isolated and used as macroinitiators for further polymerization with the same or other monomers to form block copolymers. PS/PMMA block copolymers were synthesized from polystyrene – (PS-) with active aromatic phosphorous end groups as macroinitiator. The yield of polymerization of styrene and methl methacrylate with tetraphenyl biphosphine in bulk and the yield of block copolymerization of styrene and methyl methacrylate (MMA) with PS-macroinitiator in bulk were determined in relation to the wavelength of UV light and on the ratio of initiator and monomer. The molecular weights of block copolymers increased with the increase of the reaction time. It was found out that polymerization of styrene and methyl methacrylate with tetraphenyl biphosphine gives higher yields at the wavelength of 366 nm than at 302 nm. On the other hand, block copolymerization of the PS - macroinitiator and methyl methacrylate gives much higher yield at 302 nm than at 366 nm in spite of shorter polymerization time. In the first step initiator tetraphenyl biphosphine decomposed under the influence of UV light into two primary Ph2P· radicals which are capable to initiate the polymerization. In the second step of copolymerization the macroinitiators decomposed into phosphorous radical and macroradical. The macromolecules were terminated by primary Ph2P· radicals and by the combination of two equal or different macroradicals as observed by NMR spectroscopy. The quantities of homopolymers and block copolymer in our products were determined by solid-fluid extraction.
The structure of the end-groups was determined by NMR spectroscopy. The bond length, the bond dissociation energy and the bond order of macromolecule end-groups were calculated with an electronic structure program.
W. H. BINDER and H. GRUBER
Institute of Chemical Technology of Organic Materials 162
TU-Wien, Getreidemarkt 9, A-1060 Wien, Austria
Polymers derived from the cationic ring opening polymerization of 4,5-dihydrooxazoles (“oxazolines”) show interesting physical and chemical properties with industrial applications such as coating materials, surface active agents and corrosion inhibitors for metal surfaces. Both hydrophilicity and hydrophobicity of the resulting poly(oxazolines) can be tailored by use of appropriate monomers and monomer mixtures during the polymerization process thus yielding polymers with adjustable surface active properties.
We have prepared photoactive macromonomers and polymers on the basis of polyoxazoliones for the light mediated photostabilization of micelles derived from block copolymeric micelles. In particular we focused on the p-azidophenyl moiety which is a well known photoactive group capable of crosslinking via nitrene formation. The synthesis of p-azidophenyloxazoline is accomplished in a four step sequence on a multigram scale. Subsequent living cationic polymerization enables the preparation of various homo- and block copolymers under preservation of the p-azidophenyl moiety.
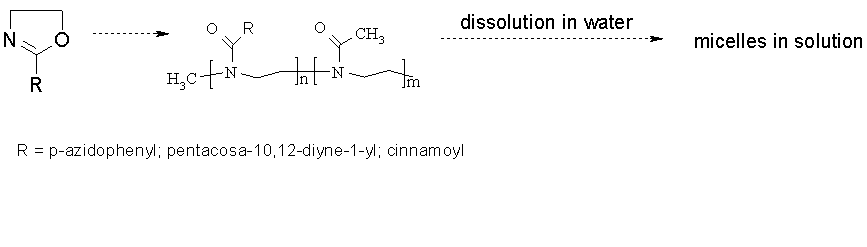
The resulting poly(oxazolines) show wide solubility in aqueous and organic solvents in dependence of their hydrophilic chain length.
The photochemical stabilization of the micelles via crosslinking is then performed by activating the photoactive groups incorporated into the block copolymers derived from oxazolines. Various other photoactive groups such as cinnamoyl- ; 1,3-diyne were also used for this purpose. The stabilization after light mediated crosslinking of the resulting micelles in aqueous solution is investigated.
OLIGOMERIZATION AND COPOLYMERIZATION OF DICYCLOPENTADIENE (DCPD)
I. PALMOVÁ † , J. KOSEK‡ , J. SCHÖNGUT † , M. MAREK‡ , K. ŠTĚPÁNEK†
† Process Engineering Department, Research & Development Center, Chemopetrol, a.s., 436 70 Litvínov - Záluží 1, Czech Republic
‡ Department of Chemical Engineering, Prague Institute of Chemical Technology, Technická 5, 166 28 Prague 6, Czech Republic
The steam cracker C5 stream is a rich resource of olefins and diolefins which can be upgraded to produce elastomers, resins and fine chemical intermediates. The most important cyclodiolefin - dicyclopentadiene (DCPD) is used for tackifying resins (55 %), unsaturated polyesters (26 %), ethylidene norbornene (14 %) and reaction injection molding (3 %). However, the largest single use for DCPD is likely to remain resins over the medium term, although the demand for DCPD used in specific products could form a significant proportion of its overall consumption, e.g., the new use of DCPD and its oligomers in the Sulfur Polymer Cement.
Two methods of oligomerization of DCPD were studied, the Diels-Alder polyaddition and the thermal (free radical) oligomerization. The oligomerization process taking place in the non-isothermal batch reactor equipped with a condenser was studied both on the laboratory and on the pilot plant scale. Products of thermal oligomerization have longer chains and better solubility in organic solvents than products of the Diels-Alder oligomerization. The copolymerization of dicyclopentadiene with maleinanhydride via the mechanism of the Diels-Alder addition was also studied experimentally. The effect of the addition of the comonomer on the resulting polymer properties (e.g., surface properties, strength, gloss, softening point, solubility) was examined.
Several methods of the analysis of the composition of reaction mixture and the properties of products were examined. For example, the capillary gas chromatography and the SIMDIS method were used for the determination of the molecular weight distribution of oligomers. The dynamics of the process was followed by the NIR spectroscopy.
The mathematical model of the Diels-Alder oligomerization in the gas-liquid system was developed and it is based on the description of the reversible reaction kinetics, gas-liquid equilibrium, local balances of mass and the energy balance. The dynamics of the process is investigated at boiling and non-boiling conditions. Kinetic parameters of the model are estimated from the dynamic evolution of concentrations of oligomers in the laboratory scale reactor.
Christelle DAVID, Delphine MALLARDE, Valérie LANGLOIS, Philippe GUERIN
Laboratoire de Recherche sur les Polymères, UMR C7581 CNRS, Université Paris XII - Val de Marne, 2-8 rue Henri Dunant, 94320 Thiais, France
A wide variety of prokaryotic organisms is used in the synthesis of poly(hydroxyalkanoates), PHAs. These polymers serve as intracellular and energy or carbon reserve materials and can be produced in large quantities by fermentation process. To date, about hundred different repeating units have been found in this PHAs family. Among these structures, 3-hydroxyacids exist with 6 to 14 carbon atoms and a large spectrum of saturated, unsaturated, linear, branched chain or functional pendant groups. One may consider biological synthesis of these bacterial polymers as a substitutite or complementary method of the macromolecular chemistry. To succeed in this approach, it will be necessary to control chemical structure through random, graft, or block copolymers and to fix molecular weight and molecular weight distribution of PHAs produced in the host organisms.
Pseudomonas oleovorans keeps a particular place due to its versatility. This strain is able to uptake and to incorporate substrates containing among other functionalized groups, terminal double or triple bonds or epoxy groups. Moreover, the composition of the produced copolyester is the strict reflection of the nutrients composition. In order to have at one's disposal polymeric materials for temporary applications and with a well controlled hydrolysis, a polyhydroxyoctanoate (PHOU) containing about 10 % of repeating units of the undecenoate type has been selected. Unsaturated bonds have been epoxidized without any modification of molecular weights. Films of both polymers have been immersed in water at two pH (pH 4 and pH 10) and the hydrolysis has been studied by different methods. Degradation of epoxidized PHOE is accelerated compared with the non modified PHOU at pH10 and thermal properties and morphology of both materials develop differently. By maintening the chemical structure of functionalized PHO, it is therefore possible to control hydrolytic degradation.
 PHOU
PHOU
GÁBOR ERDŐDI, BÉLA IVÁN
Institute of Chemistry, Chemical Research Center of the Hungarian Academy of Sciences, H-1525 Budapest, Pusztaszeri u. 59-67, P.O.Box 17, Hungary and Department of Chemical Technology and Environmental Chemistry, Eötvös University, H-1518 Budapest, P.O.Box 32, Hungary
Three-arm star hydroxyl-telechelic polyisobutylene (PIB(OH)3) with narrow molecular weight distribution (MWD) was synthetized by quasiliving carbocationic polymerization and subsequent quantitative chain end derivatization. This star polymer was cured with poly(ethylene glycol) (PEG) to form urethane linkages under suitable conditions. Randomly linked and highly ordered segmented alternating PEG-PIB amphiphilic conetworks (APCN) with high PEG contents were prepared by a new synthetic strategy. The amphiphilic nature of the resulting networks was proved by swelling in water and n-hexane. DSC measurements confirmed the existence of microphase separation in these new APCNs. Drug release experiments show constant release of a model drug from the networks over a long period of time.
Nemesio Martínez Castro and Sergio Corona-Galván
Research and Development Department, Industrias Negromex, S.A. de C.V., Carretera Tampico-Mante Km. 28.5, Altamira, Tam. 89600, México
The synthesis of functionalized polymers by anionic techniques is usually made by end-functionalization reactions, yielding polymers whose degree of functionalization is lower than 100%. The most versatile method to produce well-defined functionalized polymers is by using protected functionalized initiators. The functional groups are usually unstable to the presence of organolithium reagents, being necessary to use suitable protecting groups in the initiator,1 which should not only be stable to the anionic chain end, but they must also be easily removed at the outcome of the polymerization. Noteworthy advantages in the use of functionalized initiators include the presence of a functional group on every polymer molecule, and the easiness to produce telechelic and star functionalized homo and copolymers by termination with suitable electrophilic reagents. Here we report the use of novel protected functionalized initiators of the type R-Si-O-R-Li and R-O-R-Li in the synthesis of polybutadienes and polystyrenes by anionic polymerization in hydrocarbon solvents. The results obtained indicate that these initiators are as reactive as n-BuLi at temperatures higher than 60 0C. After acid deprotection of the polymers, we were able to get hydroxyl functionalized polystyrenes and polybutadienes, as well as star polymers by end- linking reactions, with a variety of molecular weights.
1H. L. Hsieh and R. P. Quirk, ”Anionic Polymerization: Principles and Practical Applications”, Marcel Dekker, New York (1996).
Larisa M. Timofeeva, Yulia A. Vasilieva, Natalia A. Klescheva, Galina L. Gromova and Dmitry A. Topchiev
A.V.Topchiev Institute of Petrochemical Synthesis, Russian Academy of Sciences, 29 Leninsky prosp., 117912 Moscow, Russia, E-mail:TIPS@ips.ac.ru
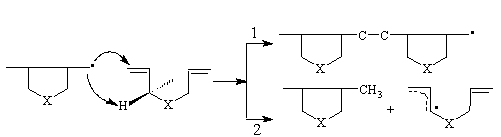
Theoretical aspects were considered of the fundamental chemical problem — including of perspective diallylamine’s (DAA’s) monomers into polymerization processes to get high molecular weight products. The latter was not realized till now because of serious limitation related to degradative chain transfer to monomer reaction with formation of stable allyl radical. But quaternary forms of DAA are known to polymerize successfully. Theoretical calculations of potential energy profiles of propagation (1) and chain transfer to monomer (2) competing reactions, simulating N,N-diallyl-N-methylamine (DAMA) as well as its protonated and quaternary forms radical polymerization in dilute solutions were performed in vacuum and in solvent within polarized continuum solvent model1 utilizing the procedure2 based on quantum chemical semiempirical MNDO-PM3 background. It was found that protonation and quaternization lead to successive increase of a -hydrogen abstraction activation energy and of difference between activation energies of competing processes, this effect being dependent on e .3 The mechanisms of phenomena is discussed. The results obtained give the possibility to predict some conditions needed to get high molecular weight polymer product based on diallylamine monomers that was just realized 4.
1. S. Miertus, E. Scrocco and J. Tomasi, J. Chem. Phys. ,1981, 55, 117.
2. G.E. Chudinov, D.V. Napolov and M.V. Basilevsky , Chem. Physics, 1992, 160, 41.
3. L.M. Timofeeva, Yu. A. Vasilieva, N. A. Klescheva and D.A. Topchiev, Russ. Chem. Bull., 1999, 865.
4. Yu. A. Vasilieva, N. A. Klescheva, G. L. Gromova, M. P. Filatova, A. I. Rebrov, E. B. Kryt’ko, L. M. Timofeeva and D.A. Topchiev. To be published.
Yulia A. Vasilieva, Natalia A. Klescheva, Galina L. Gromova,
Larisa M. Timofeeva and Dmitry A. Topchiev
A.V.Topchiev Institute of Petrochemical Synthesis, Russian Academy of Sciences, 29 Leninsky prosp., 117912 Moscow, Russia, E-mail: TIPS@ips.ac.ru
Diallylamines radical polymerization is known to control by degradative chain transfer to monomer reaction with formation of stable allyl radical and to characterize by low process rate and small molecular weights of oligomeric products.1 We showed by theoretical studies that the existence of the majority of the reagents in protonated form is the necessary condition to get high molecular weight polymer in radical polymerization of diallyl monomers, N,N-diallyl-N-methylamine (DAMA) in particular.2 Following to this idea, we chose the equimolar mixture DAMA—TFA (trifluoroacetic acid) in dilute solutions and the salt DAMA× TFA (synthesized for the first time) in solutions like polymerizing systems, DAMA being in protonated form in these systems according to NMR 1H data.3 Radical polymerization of the mixture DAMA—TFA and the salt DAMA× TFA was carried out in water solutions at various conditions. Fairly high molecular weight polybase-polyamines were synthesized for the first time at usual conditions: MM » 30000, polymerization of the mixture at 30° C in presence of ammonium persulphate (PSA) and MM » 50000, 20° C, photo-initiation, PSA; using of the monomer salt allowed to increase the initial monomer concentration and lead to the greater values of MM as it should be expected. 3 Signals of the end vinyl group in NMR 1H spectrum indicates that protonated allyl radicals are included into polymerization process and thus the degradative chain transfer transforms into the effective one. The mechanism of polymer molecular mass control was established and the possibility of regulation of process rate was showed.
A. BÖKERa, C.K. OBERb, K. REIHSc and R. STADLERa(†)
a Makromolekulare Chemie II, Universität Bayreuth, Universitätsstraße 30, 95440 Bayreuth, Germany
b Materials Science & Engineering, Cornell University, 327 Bard Hall, Ithaca, NY 14853-1501, USA
c Bayer AG, Zentrale Forschung, Oberflächenphysik, 51368 Leverkusen, Germany
In order to develop low surface energy materials it is a common approach to introduce fluorinated groups into a polymer, thus creating fluorine rich surfaces that are highly hydro- and oleophobic. Generally thin films of these products are used as non-stick coatings. Also blending of fluorinated block copolymers with homopolymers is well known to lead to dramatic changes of the surface properties of the homopolymer as the fluorinated block segregates towards the polymer/air interface, decreasing the surface energy remarkably1.
As common coatings only show one specific surface property that can not be influenced by subsequent treatment, a subject of recent interest was to synthesize fluorinated side chain block copolymers that show heat-induced selective cleavage of the fluorocarbon side chains without degradation of the hydrocarbon backbone, switching the surface properties from hydrophobic to more hydrophilic, which leads to new methods of laterally structuring and functionalizing polymer film surfaces by creating hydrophilic patterns on formerly hydro- and oleophobic surfaces.
Monodisperse poly(styrene-b-fluorinated side chain) block copolymers were synthesized by sequential anionic polymerization of styrene and isoprene, followed by specially designed polymer analogous reactions (i.e. oxidative hydroboration, reaction between the resulting alcohol and the corresponding perfluorinated acid chloride)2. By variation of the block copolymer composition and the length of the fluorinated side chain, the effects of the chemical structure on the surface composition within the upper 100 Å of thin films of the pure materials and their blends with polystyrene were examined by X-ray Photoelectron Spectroscopy (XPS), Time-of-Flight Secondary Ion Mass Spectrometry (TOF-SIMS) and contact angle measurements.
In addition to that, the thermal stability (i.e. the selective cleavage of the fluorocarbon side chains) of the fluorinated block copolymers was investigated by Thermogravimetric Analysis (TGA) and Thermodesorption Mass Spectrometry (TDMS).
References
(1) Iyengar, D.R.; Perutz, S.M.; Dai, C.A.; Ober, C.K.; Kramer E.J.; Macromolecules, 1996, 29, 1229
(2) Wang, J.; Mao, G.; Kramer, E.J.; Ober, C.K.; Macromolecules, 1997, 30, 1906-1914
T. Breiner , C. Frenz, H.-W. Schmidt
Makromolekulare Chemie I, Universität Bayreuth
In the last decades extensive studies have been carried out concerning the morphology of block copolymers based on well known monomers like styrene, methylmethacrylate and butadiene. Similar, side-chain liquid-crystalline polymers have been a subject of intensive chemical research. In our current work we aim to link both fields of research, by synthesizing blockcopolymers with liquid crystalline azobenzene side chains. First investigations in this new field was done by E.L Thomas and coworkers, who attached such groups to an isoprene backbone in a polystyrene matrix.
The Poster presents different homopolymers and blockcopolymers synthesized by living anionic technique. As strategies to obtain the final polymers a polymeranalogous reaction on blockcopolymers. In one system block copolymers of the type poly(styrene)-block-poly(butadiene) were synthesized with high degree of 1,2-polydiene content, in analogy to Gronski et. al. The polydiene-block was functionalized by hydroboration reaction to generate an poly(1-butene-4-ol) block, which can be used for further polymer analogous reactions.
The second system consists of, poly(butadiene)-block-poly(hydroxyethyl-methacrylate) which was synthesized by sequential anionic polymerization of butadiene and the protected hydroxyethylmethacrylate monomers. T-butyl-dimethyl-silyl as a rather stable protection group was selected. This group has the advantage that the protected as well as of the unprotected block copolymer can be investigated.
Various azobenzene acid chlorides were synthesized and attached to the backbone by esterification. The resulting polymers were characterized by DSC, TGA and SEC measurements. The morphologies were investigated by TEM and AFM measurements.
Katja Loos, Reimund Stadler(V )
Makromolekulare Chemie II, Universität Bayreuth, Universitätsstr. 30, 95440 Bayreuth, Germany
A potential need exists for the development of materials that show enhanced biocompatibility and bio degradability. By combining the unique properties of saccharides in biological systems and the good chemical and thermal stability of synthetic polymers, such materials can be designed. For example lactobionamide modified polystyrene already proved feasible to cultivate hepatocytes in vitro [1].
By using maltoheptaose as saccharide unit a moiety is attached that serves as a biological recognition pattern for the enzyme potato phosphorylase [2]. Therefore (,14) connected glucopyranoses can be grafted from by enzymatic polymerization. With this technique we were able to synthesize amylose modified polysiloxanes [3], silica particles [4] and polystyrenes [5] even though the starter molecules were not soluble in the medium of polymerization.
The major advantage of this polymerization procedure is the very narrow polydispersity that is obtained by the enzymatic process. Furthermore the enzyme is easy to obtain from potato tubers and tolerates not only a lot of different molecules bound to the initiating species (maltooligomers) but also a certain amount of organic cosolvents. By combining enzymatic polymerization with anionic synthesis, materials with very narrow polydispersity can be obtained.
Taking into account the Kuhn length of amylose (30 nm), the synthesized hybrid materials can be considered as rod-coil copolymers and because of the narrow polydispersity found in the block copolymers this system is very attractive to study the solution and bulk behavior of this copolymer class.
References
[1] A. Kobayashi, T.Akaike, K. Kobayashi, H. Sumitomo, Makromol. Chem. Rapid Commun. 7, 650 (1986)
[2] G.T. Cori, C.F. Cori, J. Biol. Chem 135, 733 (1940)
[3] V. v. Braunmühl, R. Stadler, Macromol. Symp. 103, 141 (1996)
[4] K. Loos, V. v. Braunmühl, R. Stadler, Makromol. Chem. Rapid Commun., 18, 927
(1997)
[5] K. Loos, R. Stadler, Macromolecules, 30, 7641 (1997)
K. Markgraf, Th. Goldacker, V. Abetz, R. StadlerU
Macromolecular Chemistry II, University of Bayreuth, 95440 Bayreuth, Germany
Block copolymers are well-known for their self-assembly into different crystal-like morphologies on a mesoscopic length scale. In comparison to binary block copolymers, ternary triblock copolymers show a much higher number of morphologies, depending basically on composition, topology and relative interactions between the different blocks. Blending of block copolymers can lead to periodic superlattices in which microdomains are formed by molecularly mixed blocks of the different blend components. Using a blend of lamellar polystyrene-block-polybutadiene-block-poly(tert-butyl methacrylate) triblock copolymer SBT with a lamellar polystyrene-block-poly((tert-butyl methacrylate) diblock copolymer st, recently the formation of a periodic non-centrosymmetric lamellar superlattice could be proven [1]. A sequence of layers ...SBTtsSBTts... could be justified by means of transmission electron microscopy and was further confirmed by small angle X-ray scattering. This is a new result since periodic non-centrosymmetry is impossible to occur for both of the blend components in the pure state. In this blend entropic forces favor the fomation of mixed microdoamins of S with s and T with t, respectively. An alternate strategy to form superlattices of two different microphase separated block copolymers with molecularly mixed microdomains takes advantage of favorable enthalpic interactions between different block components. For this puropose polystyrene-block-polybutadiene-block-poly(tert.-butyl methacrylate) triblock and poly(2-vinylpyridine)-block-poly(cyclohexyl methacrylate) diblock copolymers vc have been synthesised via sequential anionic polymerisation. Saponification of the poly(tert.-butyl methacrylate) block of the triblock copolymer results in a polystyrene-block-polybutadiene-block-poly(methacrylic acid) triblock copolymer SBA. Here mixed microdomains can be expected for A with v due to hydrogen bonding between the acidic and basic functionalities, and also S and c are miscible as known from their corresponding homopolymer blends.
The synthesis and molecular characterization of the materials will be presented together with the morphological characterisation of their blends.
References:
1. Th. Goldacker, V. Abetz, T. Stadler, I. Eurkhimovich, L. Leibler, Nature 1999, 398, 137.
H. Schmitz, V. Abetz, R. StadlerU
Macromolecular Chemistry II, University of Bayreuth, 95440 Bayreuth, Germany
Blends of microphase separated block copolymers and random copolymers are used to study the influence of a phase boundary on the miscibility of one block component with the random copolymer. Of special interest is a system, which shows a lower critical solution temperature (LCST).
Blends of polystyrene-block-poly(methyl methacrylate)-block-polystyrene triblock copolymers and poly(n-butyl acrylate)-stat-poly(cyclohexyl acrylate) random copolymers are prepared. Polystyrene-block-poly(methyl methacrylate)-block-polystyrene block copolymers show microphase separation for sufficiently high degrees of polymerisation. Poly(cyclohexyl acrylate) is miscible with polystyrene, while poly(n-butyl acrylate) shows only miscibility with polystyrene, if the degree of polymerisation is very low [1].
Miscibility of the random copolymer with the polystyrene block depends strongly on the content of cyclohexyl acrylate in the random copolymer . Within a limited composition range of the random copolymer LCST behaviour is observed.
Miscibility and morphological properties of these systems are investigated by transmission electron microscopy, atomic force microscopy, differential scanning calorimetry and dynamic mechanical testing.
The random copolymers were synthesized via atom transfer radical polymerisation (ATRP) and free radical polymerisation. The block copolymers were synthesized via sequential living anionic polymerisation [2].
References:
1) Schubert, D.W., Abetz,V., Stamm, M., Hack, T., Siol, W., Macromolecules 1995, 28,2519
2) Auschra, C., Stadler, R., Voigt-Martin, I.G., Polymer 1993, 34, 2081
U. M. WAHNER, R. BRUELL, H. PASCH, R. SANDERSON, H. G. RAUBENHEIMER
University of Stellenbosch, Institute for Polymer Science, Private Bag X1, 7602 Stellenbosch, South Africa
Odd alpha olefins e.g. 1-pentene are products of the conversion of coal via the Fischer Tropsch process, their bulk scale production is unique to South Africa (SASOL). At present, there is no commercial application for odd alpha olefins, although even alpha olefins are used as co-monomers for linear low-density polyethylene (LLDPE).
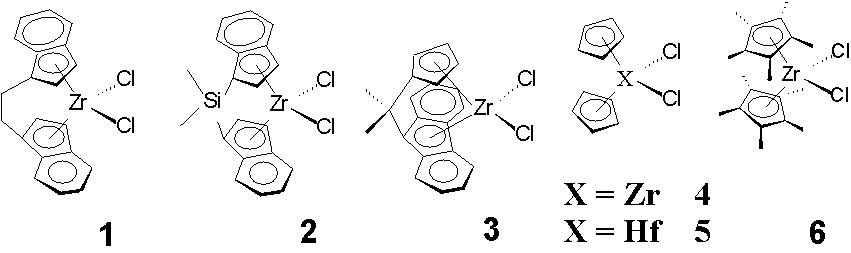
The homopoly- and oligomerisation of 1-pentene with the metallocenes 1 - 6 and methylaluminoxane (MAO), at room temperature and at 60° C, have been studied. The use of the ansa bridged metallocene 1 lead to the formation of a highly isotactic polymer. The polymer obtained with the bridged catalyst 2 had a lower degree of tacticity. The use of 3 lead to the formation of a syndiotactic oligomer, and using the metallocenes 4 - 6, atactic oligomers were obtained. The degree of polymerisation was highly dependent on the metallocene catalyst. Products ranging from the dimer of 1-pentene to poly-1-pentene with an average molar mass Mn = 5100 g/mol were formed.
The oligomers were characterised by 1H-NMR-, 13C-NMR-spectroscopy, GPC and DSC, where applicable. The 1H-NMR spectra of the samples were analysed with regard to functional groups and these attributed to different chain termination processes. A MALDI-TOF spectrum of low molar mass poly-1-pentene could be recorded.
C.J.CROOK, A.J.RYAN
Department of Chemistry, Sheffield University, Brookhill, Sheffield, S3 7HF, ENGLAND
Adhesives for porous materials (such as paper and board) and non-porous materials (such as polymer films) present different needs in terms of the physical properties of the adhesives used. The use of reactive block copolymers is desirable as they combine the 100% solids approach of hot melts with the contact adhesion mechanism of emulsion and solution based systems. The microphase separation of the copolymer gives high green strength (i.e. initial adhesion before cure) as it can be formulated to sit at the order-disorder transition and so give maximum energy dissipation.
In order to synthesise these reactive copolymers a toolbox approach has been employed whereby a series of dihydroxy functional polymers are obtained of varying chemical nature and of varying molecular weight.
The first step in the process it to react a dihydroxy functional polymer with a large excess of diisocyanate. This effectively converts the hydroxy ends to isocyanate ends. The excess reagent is then removed by thin-film distillation. This process is regularly practised in the polyurethane and adhesive industry.
The second stage involves the reaction of diisocyanate terminated polymer with another dihydroxy functional polymer to form a multiblock copolymer with an isocyanate group at each end. The adhesive can then be processed as a hot melt. Long-term creep stability comes through in situ cross-linking resulting from the reaction of isocyanate with atmospheric moisture.
The benefit of this approach is that, from a relatively small selection of polymers, a large variety of copolymers can be synthesised, e.g. only 6 starting polymers will yield a possible 15 copolymers. This means that is possible to choose a pair of polymers to suit the particular application for which the adhesive is required, avoiding problems of incompatible substrates.
The polymers used are a mixture of commercial samples and samples which have been prepared in the lab by living anionic polymerisation. All polymers are characterised by 1H and 13C NMR, GPC, and MALDI-TOF mass spectroscopy.
The resultant adhesives are to be characterised by standard industrial tests, rheology, microscopy, temperature resolved SAXS, and the adhesive bond failure mechanism will be studied using a specially designed pull-off technique.
Natalie Regnier, ETCA
are on the one hand to extend the lifetime in extreme environments and on the other hand to reduce costs by simplifying the manufacture of parts.
Currently and for structural applications, epoxy resins represent the basis of an important number of organic matrices generally strengthened with glass or carbon fiber due to their remarkable adhesive properties but also to the great variety of products whose compositions have been adapted for all kinds of applications. In the epoxy resin the aliphatic chain that links the oxirane group to the aromatic cycle is not intrinsically thermostable. The thermal limit of the best adhesives remains therefore under 180°C for a long duration use. Nowadays new thermal and mechanical requirements for aeronautic industry lead to the development of new candidates.
Thus, materials based on cyanate resins seem to present a good compromise between current epoxy resins and resins for "high temperatures" as bismaleimide resins. These last years, manufacturers have developed a thermostable epoxyde composite for the conception of helicopters. A new formulation composed of epoxy and cyanate monomers has been developed to reach the performance of self-adherence.
This study presents the influence of temperature on the damage and mechanical properties of epoxy and epoxy-cyanate resins carbon fibre composites. Matrix undergo chemical decomposition on thermal treatment with the loss of volatile products and induce a mechanical properties failure. We propose a relation between chemical structure and mechanical properties (ILSS, tan d ) of these two epoxy composites.
Saverio Russo, Maria Elisabetta Cosulich, Sara Pasquale and Pietro Canepa
Dipartimento di Chimica e Chimica Industriale – Università di Genova, Italy
Samples of hyperbranched aramids, originated either from AB2-type monomers or from reactant pairs A2+B3, A3+B4, have been synthesized under different reactions conditions and used as a-amylase supports. A high efficiency of protein immobilization has been observed, the binding capability being almost an order of magnitude higher than the commonly used insoluble supports. Each preparation of the immobilized enzyme has been evaluated considering its bioactivity retention vs.time, as well as stability under various experimental conditions. Enzymatic activity has been evaluated in terms of enzyme affinity for the substrate and catalytic efficiency.Our data attest that immobilization process leads to the production of three different types of adducts with distinct enzymatic patterns. The final run of investigations has been conducted with real rice boiled water as substrate of these adducts and the degree of biodegradation has been evaluated by chromatographic analysis. The effectiveness of the bioreactor is influenced by the time of contact with the immobilyzed enzyme: in this attempt we have followed the hydrolysis of the starch and the production of oligosaccarides and maltodextrines by stirring 1 liter of rice boiled water in the presence of 100mg of adduct for 90min. On the basis of our results it may be inferred that hyperbranched aramids are suitable supports for protein immobilization. The availability of these dendritic structures by simple synthetic preparations can open new perspectives for the development of finely tuned enzyme-based derivatives with pre-defined binding affinity, catalytic capability and structural stability. Molecular modelling of the above hyperbranched structures has been very useful in the interpretation of subtle differences in behaviour among the various adducts.