1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
SMALL-ANGLE NEUTRON SCATTERING FROM ADSORBED POLYMER LAYERS
S M KINGa, P C GRIFFITHSb & T COSGROVEc
aISIS Facility, Rutherford Appleton Laboratory, Chilton, Didcot, OX11 0QX, UK
bChemistry Department, Cardiff University, PO Box 912, Cardiff, CF10 3TB, Wales
cSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK
One of the areas in which small-angle neutron scattering (SANS) has had a profound impact over the last twenty years is in the investigation of polymers adsorbed at the solid/liquid and liquid/liquid interfaces. This is of course also an area of considerable commercial importance. Adsorbed polymers are used to control the rheology and colloidal stability of cosmetic and pharmaceutical preparations, paint and agrochemical formulations, and even foodstuffs. Polymer adsorption is also important in the biological arena where, for example, it mediates in the cellular adhesion process. Control of the structure of an adsorbed layer is thus a key priority of the colloid scientist. Equally important, however, is the ability to probe the arrangement of the adsorbed polymer molecules and to extract quantitative information from what might well be a complex, multi-component, system.
SANS, in combination with the principle of deuterium contrast variation, is uniquely placed to provide this information. As a scattering technique it provides directly (unlike rheological and viscometric measurements) microstructural information about bulk systems (unlike ellipsometry and reflectivity) on shorter length scales and with better resolution than is possible with light scattering.
This lecture will give an evolutionary review, with examples, of the different approaches that may used to interpret SANS data from polymer molecules adsorbed in colloidal dispersions.
A few remarks on classical problems of scattering by polymers solutions and mixtures
Henri Benoît* and Gérard Jannink°**
*Institute Charles Sadron, 6, rue Boussingault, 67083, Strasbourg
France
**Léon. Brillouin Laboratory,C.E. Saclay, 91191 Gif-sur-Yvette, CEDEX, France
The problem of scattering by multicomponent systems has been the object of many papers. Here it will be shown that the scattering of these systems can be split into two terms, one corresponding to the compressibility; the other depending only on the fluctuations of the number of molecules of the different constituents. In the practice of neutron scattering on samples far from critical conditions, the compressibility term is completely negligible and its contribution is suppressed with the incoherent scattering subtraction. In a second part a simple demonstration of the Random Phase Approximation theorem is proposed This demonstration explains the title of this theorem since the result is obtained by changing the averaging or one correlation term. This explanation is very simple and makes this theorem more understandable for the non-specialists. It should justify its use more often.
Linear, branched and hyperbranched macromolecules in dilute solution
Fabio Ganazzoli
Dipartimento di Chimica - Politecnico di Milano, via L. Mancinelli 7 - Italy
E-mail: Fabio.Ganazzoli@polimi.it
Web page: http://dept.chem.polimi.it/home_page/ganazzoli
Branched and hyperbranched molecules such as regular stars and dendrimers were synthetized in recent years with unprecedented control over molecular architecture. Here, I shall report some theoretical results obtained for these macromolecules using the Gaussian Self–consistent approach. I first discuss the Q state. Both star polymers and dendrimers are predicted to show a depression of the Q temperature compared to linear chains, and a larger size than predicted with the random-walk model when the number of star arms or the dendrimer generation are large. However, while stars asymptotically achieve the same Q temperature as linear chains, dendrimers are predicted to show an increasingly lower Q value with increasing generation. These features are explained by residual interactions among the monomers, related in turn to the molecular topologies.
Then I consider the good-solvent expansion of the molecule. This expansion turns out to be concentrated near the central core, while the star arms or the dendra, sub-dendra etc, are basically independent and somewhat segregated, so as to simultaneously minimize both intra- and inter-arm (or intra- and inter-dendron) repulsive interactions. Concerning the intramolecular dynamics, I briefly discussion the spectrum of relaxation times and the dynamic structure factor. Its first cumulant, in particular, allows estimating the error of the preaveraging approximation for the hydrodynamic interaction.
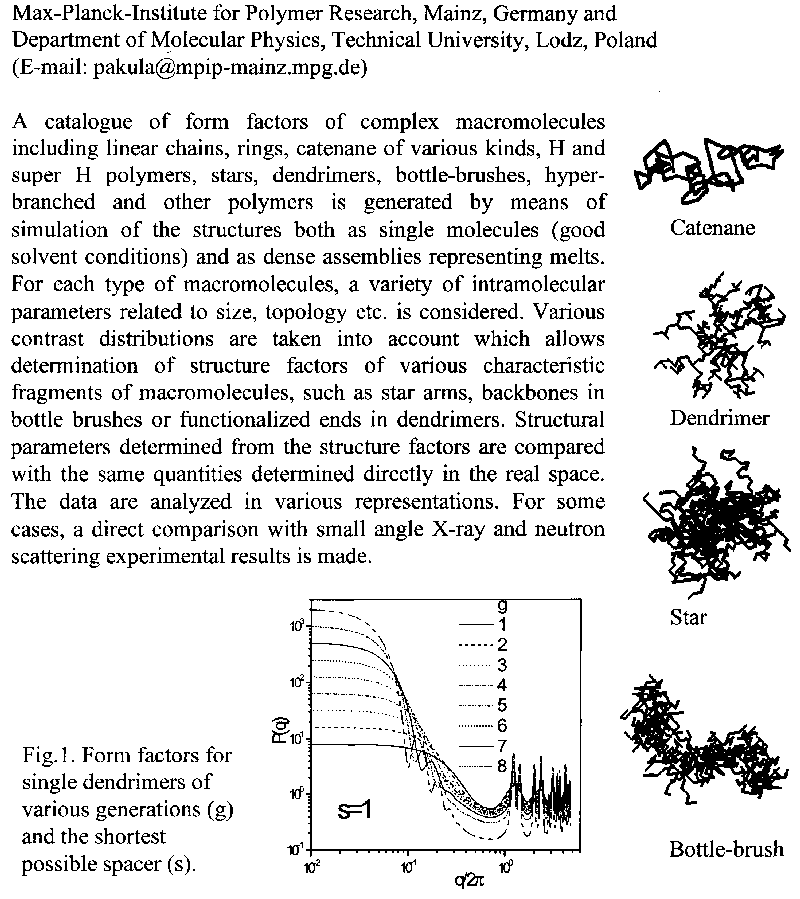
Structure Factors of Complex Macromolecules
T. Pakula
STRUCTURAL STUDIES OF COLLOIDAL AND POLYMER SYSTEMS UNDER THE INFLUENCE OF SHEAR
Kell MORTENSEN
Danish Polymer Centre, Risø National Laboratory, DK-4000 Roskilde, Denmark
Many polymer and colloidal systems are exposed to major external shear and strain during both manufacturing and in the applications. It is therefor of great importance to study experimentally the structural and mechanical response of model systems in situ during shear and stress. A commercial rheological instrument, the Rheometrics RSA-2, has been modified for such combined studies of rheological and structural parameters. The rheological parameters were obtained in the oscillatory shearing mode, and structure was obtained by small-angle neutron scattering. Another homemade Couette-shear device has been constructed for constant shear experiments. Rheo-SANS experiments based on these devices have been performed on a variety of polymer(1) and colloidal(2) materials. The studies include investigations of the changes in texture of the ordered phase of di- and triblock copolymer melts and gels upon application of shear, changes in the order-disorder phase transition and shear induced order-order transitions. transitions.
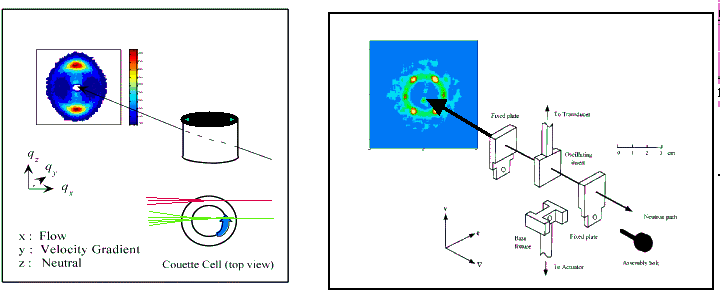
Fig. 1 Couette and parallel plate shear devises used in situ with small angle neutron scattering experiments. The scattering examples are those of shear induced nematic phase of aqueous system of C10E3 (2) and gel of SEBS (1).
THE DYNAMICS IN MICROPHASE-SEPARATED BLOCK COPOLYMER SYSTEMS
C.M. PAPADAKISa, F. RITTIGa, J. KÄRGERa, K. ALMDALb,
K. MORTENSENb, P. ŠTĚPÁNEKb
aFakultät für Physik und Geowissenschaften, Universität Leipzig, Linnéstr. 5, D-04103 Leipzig, Germany
bRisø National Laboratory, P.O. Box 49, DK-4000 Roskilde, Denmark
cInstitute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, Heyrovsky sq. 2, CZ-162 06 Praha 6, Czech Republic
The dynamic processes taking place in microphase-separated block copolymer systems are still under discussion. We studied the influence of the morphology on the dynamics in the hexagonal, the gyroid and the cubic state in poly(ethylene propylene)-poly(dimethylsiloxane) diblock copolymers. By combination of small-angle neutron scattering [1], dynamic light scattering (DLS) [2] and pulsed field gradient (PFG) NMR [3], we could for instance determine the anisotropy of polymer diffusion in the hexagonal state. Also, in the hexagonal and the cubic morphology we detected polymers which are not bound to cylinders/micelles but diffuse through the matrix. One of the samples forms different ordered phases as a function of temperature [1] allowing us to study the influence of structural changes on the dynamics in the same sample.
Another system under investigation is a ternary blend of two homopolymers and the corresponding diblock copolymer which forms a bicontinuous microemulsion. PFG NMR allows us to monitor the changes of the homopolymer diffusivities compared to the bulk values, which are due to the bicontinuous structure and to partial mixing.
[1] C.M. Papadakis, K. Almdal, K. Mortensen, M.E. Vigild, P. Štěpánek, J. Chem. Phys. 111 (1999) 4319.
[2] C.M. Papadakis, K. Almdal, K. Mortensen, F. Rittig, G. Fleischer, P. Štěpánek, Eur. Phys. J. E 1 (2000) 275. C.M. Papadakis, K. Almdal, K. Mortensen, F. Rittig, P. Štěpánek, Macromol. Symp. 149 (2000) 99.
[3] F. Rittig, J. Kärger, C.M. Papadakis, G. Fleischer, P. Štěpánek, K. Almdal, Phys. Chem. Chem. Phys. 1 (1999) 3923.
LIGHT SCATTERING INVESTIGATIONS OF ASSOCIATION PHENOMENA IN SALINE CARRAGEENAN SOLUTIONS: MOLECULARITY OF SALT- AND TEMPERATURE-INDUCED CONFORMATIONAL TRANSITIONS
H. REYNAERSa, S. PAOLETTIb, K. BONGAERTSa, F. CUPPOa
aCatholic University of Leuven, Laboratory of Macromolecular Structural Chemistry, Celestijnenlaan 200F, B-3001 Heverlee, Belgium
bDepartment of Biochemistry, Biophysics and Macromolecular Chemistry (BBCM), University of Trieste, Via L. Giorgieri 1, I-34127 Trieste, Italy
The conformational transition of kappa- and iota-carrageenan in different salt solutions under non-gelling conditions has been studied by WALLS and LALLS. An upward curvature of the reduced scattering intensity as a function of the polymer concentration in conditions of conformational ordering has been interpreted in terms of a reversible intermolecular association. Consequently, the Open Association Model (OAM) of Elias has been applied for the elaboration of the data. Temperature and concentration of the supporting 1:1 electrolyte were chosen so as to span across the disorder-to-order conformational transition. All results point to the single helix as the fundamental ordered conformation of both kappa- and iota-carrageenan in aqueous solution. This conclusion appears to hold in a large number of cases, including polymer samples of different molar mass, in the presence of different counter-ions and co-ions, and at different temperatures. The single-helical structure is the building block of a higher-level structure, that stems from the association of at least two of such fundamental units, according to a topology that can not be determined by light scattering data only. Special attention will be given to the discussion of association phenomena induced by Cs+ ions in NaI/CsI mixed salt solutions.
SANS and Light Scattering from BSA-PDADMAC Coacervates”
H.B. Bohidar1 , J. Lal2 and P.L. Dubin3
1School of Physical Sciences, J. N. University, New Delhi-110067, India (bohidar@usa.net).2IPNS, Argonne National Laboratory, IL 60439 USA (jlal@anl.gov).3Chemistry Department, Indiana-Purdue University, IN 46202, USA (dubin@chem.iupui.edu)
Protein-polyelectrolyte coacervates comprise a novel state of macromolecular fluids. These optically clear phases contain protein in excess of 150 g/L concentrations which are normally not homogeneously sustainable in aqueous solutions but which are characteristic of those within cells. They display very large shear viscosities, but exhibit protein diffusivities only an order of magnitude below those in dilute protein solution. Here, we probe the microscopic structure of coacervates prepared through pH-induced complexation of bovine serum albumin with the strong polycation poly(dimethyldiallylammonium chloride) using small angle neutron scattering and static and dynamic light scattering. The results taken together reveal that the coacervate phase is a solution-like state in which homogeneous fluid-like domains coexist with denser and more nearly charge-neutralized domains which inhibit local protein diffusion and confer transient network rheology .
Selection of Single Scattering from Multiple Scattering Systems by 3D-Cross-Correlation.
2. Concentrated Polymer Solutions
Lisa Aberle*, Malte Kleemeier, Otto-Dietrich Hennemann
Fraunhofer-Institute for Manufacturing and Advanced Materials
(IFAM)
Wiener Str. 12, 28359 Bremen, Germany; e-mail: ab@ifam.fhg.de
Walther Burchard
Institute of Macromolecular Chemistry, University of Freiburg,
Stefan Meier Str. 31, 79104 Freiburg, Germany; e-mail: burchawa@ruf.uni-freiburg.de
Characterization of colloidal and macromolecular systems become complex in concentrated solution. The solutions are often turbid and display pronounced multiple scattering. Recently a fairly simple set-up has been developed by one of us that allows selection of the single scattering component by 3D cross-correlation. Application to a colloidal system consisting of polystyrene latices in water revealed that correct particle scattering factors and correct hydrodynamic radii are indeed obtained. Neglect of multiple scattering in dynamic light scattering led to systematic errors, and wrongly indicated a decrease of the hydrodynamic radii as the concentration was increased.
In the present contribution the 3D cross-correlation is extended and applied to polymer solutions consisting of the highly branched, water soluble amylopectin (waxy maize) in concentrations up to 20% (w/v). The molecular parameters of this molecule are Mw = 70´ 106 g/mol, Rg = 253 nm and Rh = 316 nm. Surprisingly, and in contrast to results from colloids, no detectable change in the time correlation function and the corresponding mutual diffusion coefficient was observed in spite of marked multiple scattering. The present measurement appear to confirm previous observations made with latex particles of different size. Accordingly multiple scattering seems to loose its influence on the time correlation function when the particles increase in size. This effect could be the result of strong forward scattering connected with large particles. Monte Carlo are in progress that will clarify this question.
Structure Development in Polyaniline Films during Electrochemical Polymerization: Time-Resolved Rayleigh Scattering Photometry
Tadao KOTAKA, Hikaru OKAMOTO*
Polymeric Materials Engineering, Toyota Technological Institute,
Hisakata 2-12-1, Tempaku, Nagoya 468-8511, Japan
*Present address: IMRA Material R&D Co., Kariya 448-0021, Aichi, Japan
ABSTRACT: Structure development in polyaniline (PAn) films was followed during the electrochemical polymerization in 1 M HCl solution via a constant potential mode at + 0.76 ± 0.03 V and pH = 0.2 by using several in-situ and/or ex- situ techniques including UV-visible spectrometry, chronoamperometry, Rayleigh scattering (LS) and atomic force microscopy (AFM).
A crucial problem in the LS analysis of PAn film is that the PAn chains assume a greenish oxidized (emeraldine) form under the polymerization potential of +0.75 V, thereby making in-situ LS measurement impossible, and only at -0.2 V become a colorless reduced (leucoemeraldine) form. This difficulty was circumvented by switching the potential back and forth from +0.75 to -0.2 V, at which the polymerization temporarily stopped but the deposited PAn film was colorless and the LS data could be collected.
As the polymerization proceeded from the initial nucleation stage I (where the polymerization current density varies as I t0 ) to the intermediate stages IIa and IIb ( I t3 t2 ) and the final stage III (I t4 ), the gross morphology changed from grainy to fibrillar texture. Since the polymerization current density is proportional to the mass of PAn deposited and LS intensity Is(q) is proportional to the square of the mass deposited, the LS invariant Q Is(q) t8 in the final stage III of polymerization.
From the AFM observation, the fibrils were turned out to be aggregates of the grains with the size unchanged from that formed in the early stage. Thus difference between the grainy and fibrillar structures is due to the difference in the state of aggregation of the initially formed grains. In the later stages, however, the fibrils began to form branches, accelerating the rate of polymerization. The branched fibrils grew toward vertical direction keeping their horizontal structural size virtually unchanged.
TEMPERATURE-INDUCED CONTRAST VARIATION AS A TOOL FOR CHARACTERIZING ANISOTROPIC POLYMER MICROSTRUCTURES
J. D. BARNESa, R. KOLBa
aJDB Science, LLC, 7710 Chatham Rd, Chevy Chase, MD 20815 USA
bExxon-Mobil Engineering R&D Laboratory, Annandale, NJ, USA
In earlier work we found that the differences in thermal expansion coefficient between the amorphous and the crystalline components of the microstructure in certain semicrystalline polymers[1,2] caused their small-angle scattering patterns to change strongly as a function of temperature over a modest range of temperatures well below their annealing range. These results provided enough information to permit us to separate the form factor of the lamellar stack microstructure from that due to voids and other kinds of density fluctuations.
We have extended this work to encompass materials whose specimen symmetry is orthorhombic. This required that we collect the full 3-dimensional SAXS and WAXD patterns from these materials. The specimens studied here were a commercial HDPE product and a P4MP material that was formed by channel die extrusion. Both of these materials exhibit lamellar stack morphologies, but the differing process histories cause the morphologies to take on strikingly different forms in the two cases. The temperature-induced contrast variation effect was exploited to separate the void microstructure from the lamellar microstructure in both materials.
The approaches used in this work are applicable to a wide range of materials formed from semicrystalline polymers, which are perhaps the most widely used plastic products. Synchrotron x-ray cameras greatly facilitate the acquisition of the considerable volume of data that is needed to provide information of the kind that is needed to accurately reconstruct the 3-d scattering patterns in both the SAXS and WAXD regimes.
Time-Resolved SAXS Studies on Crystallization Behavior of Poly(ethylene isophthalate-co-terephthalate)s
M. Ree*, B. Lee, T. J. Shin, S. W. Lee, and J. W. Lee
Department of Chemistry, Center for Integrated Molecular
Systems, and BK21 Functional Polymer Thin Group, Pohang
University of Science and Technology, San 31, Hyoja-dong, Pohang
790-784 South Korea (Tel) +82-54-279-2120,
(Fax) +82-54-279-3399 (E-Mail) ree@postech.edu
Isothermal crystallization and subsequent melting behaviors, as well as morphology in random copolyesters, poly(ethylene isophthalate-co-terephthalate)s were investigated by small-angle X-ray scattering (SAXS) and differential scanning calorimetry (DSC). Isothermally crystallized copolyesters reveal a multiple melting behavior in the DSC thermogram: i) melting of the primarily crystallized crystals, ii) melting of the secondarily crystallized crystals, and iii) melting of the crystals recrystallized during the heating run of the DSC measurement. Overall, the melting point of the secondarily crystallized crystals shifts to the high temperature region with increasing the time of isothermal crystallization but the other crystal melting points vary insensitively with isothermal crystallization time. From SAXS patterns measured during crystallization and melting, morphological parameters are estimated. As the crystallization time increases, the SAXS pattern moves its peak maximum to the high scattering angle region and furthermore its invariant Q decreases. The copolyesters have amorphous layer thicker than that in the hompolymer, poly(ethylene terephthalate) (PET) but their lamellar thicknesses are comparable to that of the PET. These DSC and SAXS results suggest that the secondary crystallization leads densification via fringed micelle like molecular aggregation in the amorphous layer region that is the interlayer of lamellar stacks formed by the primary crystallization. In addition, the Pohang Synchrotron Facility including SAXS and WAXD beamlines will be introduced.
[This study was supported by the Korean Ministry of Science & Technology (MOST) via KISTEP (Basic Research Grant of Nuclear Energy) and by the Center for Integrated Molecular Systems (KOSEF). The synchrotron SAXS measurements were supported by MOST and POSCO.]
THE CRYSTALLIZATION AND MELTING OF LINEAR POLYETHYLENE STUDIED BY TEMPERATURE MODULATED SAXS AND WAXD
B. GODERISa, H. REYNAERSa, M.H.J. KOCHb
aCatholic University of Leuven, Laboratory of Macromolecular and Structural Chemistry, Celestijnenlaan 200F, B-3001 Heverlee-Leuven, Belgium
bEuropean Molecular Biology Laboratory, EMBL c/o DESY, Notkestrab e 85, D-22603 Hamburg, Germany
There is evidence based on temperature-modulated differential scanning calorimetry (TM-DSC) experiments for a small fraction of temperature reversible melting and crystallization in semi-crystalline polymers [1-3]. This implies crystallization without supercooling. Classical polymer crystallization theories rely on nucleation concepts and only account for irreversible crystallization.
Temperature reversible and non-reversible events could be separated in the case of linear polyethylene during quasi-isothermal crystallization by using simultaneous temperature-modulated synchrotron SAXS and WAXD [4]. Crystallization and subsequent annealing was followed for 90 min at 126 °C while applying a temperature modulation with an amplitude of 1°C and a period of 2 min. As expected the crystal growth rate associated with the irreversible part of the crystallization decreases with increasing temperature in a cycle. Secondly, the crystalline lamellae irreversibly thicken with time. The actual crystallite thickness, however, exhibits a superimposed modulation out of phase with that of the temperature modulation. Hence, reversible melting and crystallization takes place at the fold-surface of the lamellar crystallites.
Melting was studied during heating at 1°C/min after cooling at 10°C/min. In that case a temperature modulation was superimposed with an amplitude of 2°C and a period of 3 min. Fold surface activity could be observed as well. There is a good agreement between the amount of reversibility estimated from TM-DSC and SAXS.
Finally, SAXS in combination with a temperature modulation was recognized as a convenient way to cope with Babinet's principle.
[1] I. Okazaki, B. Wunderlich, Macromolecules, 30, 1758 (1997); [2] K. Ishikiriyama, B. Wunderlich, Macromolecules, 30, 4126 (1997); [3] W. Hu, T. Albrecht, G. Strobl, Macromolecules, 32, 7548 (1999); [4] B. Goderis, H. Reynaers, R. Scherrenberg, V.B.F. Mathot, M.H.J. Koch, Macromolecules, 34, 1779 (2001)
Heterodyne and Non-Ergodic Approach to Dynamic Light Scattering of Polymer Gels: Aqueous Xanthan in the Presence of Metal Ions (Al(III))
Andrew B. Rodd 1,2, Dave E. Dunstan, DavidV. Boger 1,*, J. Schmidt 2, W. Burchard 2

Diffusing wave spectroscopy of nonergodic media: Dynamics of concentrated colloidal suspensions and gels
FRANK SCHEFFOLD, SARA ROMER, HUGO BISSIG, ANNA STRADNER, VLADIMIR LOBASKIN, VERONIQUE TRAPPE, LUCA CIPELLETTIb AND PETER SCHURTENBERGER
Physics Department, University of Fribourg, CH-1700 Fribourg,
Switzerland;
b GDPC, Université de Montpellier II, 34095
Montpellier Cedex 05, France
The application of static and dynamic light scattering to concentrated colloidal suspensions has often been considered too complicated due to strong multiple scattering. Here we show that diffusing wave spectroscopy (DWS) permits the characterization of dynamic properties of such systems on a large range of time and length scales ranging from a few Angstroms to hundreds of nanometers. We focus in particular on the study of the aggregation and sol-gel transition in concentrated colloidal suspensions. We present a new technique to overcome the problem of nonergodicity in DWS of solid-like systems.[1] Using this technique we obtain quantitative information about the microscopic dynamics all the way from an aggregating suspension to the final gel, thereby covering the whole sol-gel transition.[1,2] Moreover, wer show that we can use a fast CCD camera combined with multispeckle correlation function analysis for the DWS measurements with nonergodic systems in order to extend the accessible time scale to very long times and thus are able to study ageing phenomena.
We demonstrate that concentrated suspensions exhibit universal features during the sol-gel transition. We observe for the local (microscopic) particle dynamics an abrupt transition from free Brownian diffusion to an arrested subdiffusive motion which is clearly related to the corresponding build-up of the elastic properties of the macroscopic particle gel. We demonstrate that we can extract from the DWS data quantitative information about the viscoelastic properties of the suspensions and gels such as the elastic modulus and the frequency dependent storage and loss moduli over an extended range of frequencies, and we compare these data to those directly obtained from rheological measurements.
[1] a) S. Romer, F. Scheffold, P. Schurtenberger, Phys. Rev. Lett. 2000, 85, 4980, b) F. Scheffold, S.E. Skipetrov, S. Romer, P. Schurtenberger, Phys. Rev. E (in press)
[2] S. Romer, C. Urban, H. Bissig, A. Stradner, F. Scheffold, and P. Schurtenberger, Phil. Trans. R. Soc. London A 359 (in press)
DYNAMIC LIGHT SCATTERING: AN USEFUL TECHNIQUE TO FOLLOW GELATION AND PHASE SEPARATION IN POLYMER MIXTURES
M. C. BLANCO, D. LEISSNERa, C. VÁZQUEZ AND M.A. LÓPEZ-QUINTELA
Dept. Physical Chemistry, Fac. Chemistry, University of Santiago de Compostela, E-15782 Santiago de Compostela, Spain.
aResearch Center for Materials Science, Nagoya University, Chikusa-ku.
Nagoya 464-8602, Japan.
Gelation and phase separation (PS) are very important processes which are very interesting because they appear in many different practical applications. Therefore, it is very important to have techniques able to follow such processes. There are a number of techniques commonly used to study separately both gelation and PS, like, viscosity, confocal microscopy, etc. But in many cases gelation and PS are coupled so that it should very convenient to develop suitable techniques to be employed in such cases.
Dynamic light scattering (DLS) has not been employed so far for the study of coupled gelation/PS processes mainly because of the dificulty in the evaluation and interpretation of the data, which is very controversial. Recently, we have developped an interpretation of the DLS modes in transient gelatin gels1, which can also be succesfully applied to study more complex systems, like gelation kinetics, percolation in microemulsions, and coupled gelation/PS processes.
In this work we will describe some of the most important points of the DLS mode`s interpretation in transient gels, and this will be applied further to several examples including the microphase separation in temperature quenched geling mixtures of gelatin and maltodextrin.
1 Blanco, MC; Leisner, D; Vazquez,
C; Lopez-Quintela, MA. Dynamic light scattering in
transient reversible gels.
LANGMUIR, 2000, 16: 8585-8594.
STRUCTURE-PROPERTY RELATIONSHIPS FOR FUNCTIONALLY TAILORED POLYMER BLENDS NY TIME-RESOLVED LIGHT SCATTERING
Y. A. AKPALU
NYS Center for Polymer Synthesis, Department of Chemistry, Rensselaer Polytechnic Institute, 110 8th Street, Troy NY 12180
In this lecture, an effective multivariable measurement approach to correlate structural organization with melt phase separation and chemical structure in semicrystalline systems will be discussed. Significant improvements in technologies that govern new product generation and optimization of currently available crystal-amorphous blend products by the polymer industry depend on the ability to correlate polymer structure resulting from melt phase separation, crystallization and processing conditions with desired properties. For many applications, the microscale and nanoscale morphology determine the overall function of the particular application or device. In order to understand what controls the microscale structural organization in these systems, one needs a method to measure and separate density fluctuations arising from composition fluctuations in the melt state as well as, density fluctuations arising from the crystals and orientation fluctuations arising from the arrangement of anisotropic crystals. Light scattering is a non-destructive technique that can be used to provide this information while complementary morphological information can be obtained from polarized optical microscopy (POM). New insights into the coupling of phase transitions and the segregation of amorphous polymers revealed by time-resolved light scattering on model crystal-amorphous blends and homogenous ethylene copolymers will be discussed. The use of the light scattering techniques for high throughput measurements of specific properties of multiphase systems will also be discussed.
The Impact of Thermodynamic Interaction on the Structure and Dynamics of Isotopic and Binary Short Chain Poly(siloxane) blends as Revealed by Elastic and Quasielastic Neutron Scattering
Bernd Ewen, Max-Plack-Institut für Polymerforschung, PO 3148, D55021 Mainz, Germany; e-mail: ewen@mpip-mainz.mpg.de

THE USE OF MONTE-CARLO SIMULATIONS TO CALCULATE SMALL-ANGLE SCATTERING PATTERNS
B.P. GRADYa,b AND B.C. McALISTERb
aMax Planck for Colloid and Interface Science, Potsdam, Germany 14424
bDepartment of Chemical Engineering, University of Oklahoma, 100 East Boyd, Norman OK 73019
Monte-Carlo (MC) simulations are widely used to solve polymer physics problems. The purpose of this talk is to summarize the progress in the use of MC simulations to solve small-angle scattering problems, with specific emphasis on morphologies common to polymeric systems.
MC methods have been applied in two distinct ways to polymer systems that scatter at small angles. The first way is exemplified by scattering from isolated chains in solution, where MC simulations are used to simulate the conformation of the polymer chain and calculation of the scattering pattern is usually done directly, i.e. without Monte-Carlo methods. This type of simulation, although extremely important in polymer science, will only be briefly described and is not be the focus of this talk.
Rather, the focus of this talk will be on the use of MC simulations to calculate scattering patterns from multiphase or colloidal systems. The general procedure for this type of analysis is to place the object(s) according to some criteria, then use a MC method to select pairs of points, and finally calculate scattering intensities from these randomly selected points. The authors‘ interest in this problem was generated by the following observation: the proliferation of synchrotron sources and 2-D detectors has meant that anisotropic scattering patterns are easily measured; however methods to translate this pattern into a corresponding morphology exist only for very specific morphologies. A Monte-Carlo method could simulate scattering for any morphology, and with a suitable least-squares fitting routine, could be used to fit data. This talk will describe progress in Monte-Carlo simulations of small-angle scattering.
The simplest type of morphology, an isolated sphere, turns out to be an extremely rigorous test of simulation accuracy. Three different expressions with various levels of simplification can be used in combination with MC methods to calculate scattering from a sphere, and the simulation time is proportional to the simplifications. An anisotropic single object with random orientation (i.e. a collection of objects where interparticle interference can be neglected) is the next level of difficulty, followed by polydisperse spheres, finally followed by a collection of widely-separated anisotropic objects with preferred orientation and/or size distribution. The strategies for each of these systems will be reviewed, and, based on simulation time, we will assert that MC simulations could be used to develop a least-squares routine to fit experimental data for all of these situations except, in certain cases, the last. Five types of more densely packed systems can be considered, and even for the most simple systems, MC simulations have proven to be less successful. Projections for the future of these methods will be given.
Monitoring Kinetic Processes in Polymer Solutions with Time Dependent Static Light Scattering (TDSLS)
Wayne F. Reed
Physics Dept., Tulane University, New Orleans, La. 70118, USA
An overview of recent developments in TDSLS and auxiliary techniques will be given. This includes background theory, results and applications for:
* Automatic, Continuous Online Monitoring of Polymerization Reactions (ACOMP)
* Kinetic and structural determinations from degrading polymers
* Dissolution of dry polymeric powders
* Aggregation
* Heterogeneous Time Dependent Static Light Scattering (HTDSL) of co-existing polymer and colloid systems.
* Automated methods for equilibrium batch characterization of neutral polymers and polyelectrolytes.
The Effect of Water on the Solution State of Cellulose in the Solvent System N,N-dimethylacetamide/lithium chloride - A Reason for Inconsistent Results in Size Exclusion Chromatography (SEC)
Thomas Röder 1,3(*), Antje Potthast 1, Thomas Rosenau 1, Paul Kosma 1, Thomas Baldinger 3, Bernd Morgenstern 4, and Otto Glatter 2

P. ZIEGLERa, O. SPYCKERELLE a, B. HAIDAR a, A. VIDAL a, J. OBERDISSE b, F. BOUE b
a Institut de Chimie des Surfaces et Interfaces (CNRS), 15 rue Jean Starcky, BP2488, F-68057 Mulhouse Cedex, France, P.Ziegler@uha.fr
b Laboratoire Léon Brillouin, CEA Saclay, F-91191 Gif-sur-Yvette Cedex, France
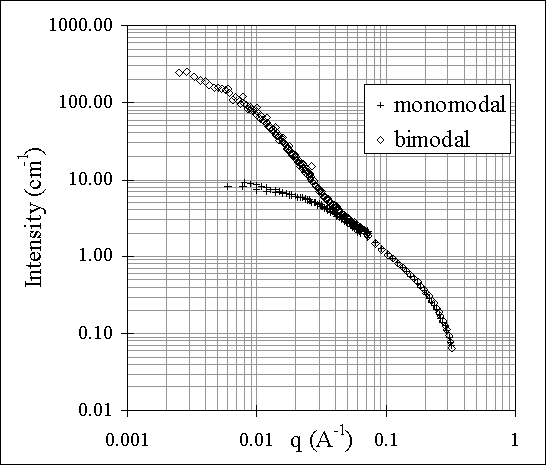
The concept of bimodal networks has been further extended to create networks with unexpected physical properties. This work will concentrate on networks of hydroxy-terminated poly(dimethylsiloxane) (PDMS) cured at room temperature. Results show that the type of chains used to form the network, the amount of cross-linking agent, the catalyst level as well as the content of short chains, in the case of bimodal networks, are critical parameters in attaining a "self-reinforcement" effect. In particular, it can be shown that bimodal networks containing around 30% (w/w) of short enough chains are substantially tougher than monomodal networks. The structure of the corresponding networks has been investigated by a small-angle neutron scattering techniques. Scattering was performed at room temperature in the swollen state with D8-Toluene to ensure sufficient contrast and all the data were treated by classical methods. By fitting the scattered intensity curves versus the scattering vector, this technique allows us to obtain different results among which typical correlation lengths of the studied network. The differences between a monomodal network based on long chains (58000 g/mol) and a bimodal network with 30 weight % of very short chains (400 g/mol) with the same long chains are illustrated on figure 1. An excess scattering is clearly observable at small q values related to another correlation length in the network.
The results presented here will be treated in terms of the relationship between network structure and the "self-reinforcement" effect observed for well defined bimodal networks.
STRUCTURE AND ORDERING KINETICS OF MICELLES IN TRIBLOCK COPOLYMER SOLUTIONS IN SELECTIVE SOLVENTS
R. BANSILa, H. NIEa, Y. LIa, K. LUDWIGa, M. STEINHARTb, C. KONAKb, J. LALc
aDepartment of Physics, Boston University, Boston, MA 02215 USA
bInstitute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, Heyrovského nám. 2, CZ-162 06 Praha 6, Czech Republic
cIPNS Division, Argonne National Laboratory, Argonne, IL-60439 USA
We have used small angle x-ray scattering (SAXS), and small angle neutron scattering (SANS) to study the micelle structure of a polystyrene (S)- block-poly (ethylene-co- butadiene) (EB)- block-polystyrene triblock copolymer, SEBS (Kraton G1650) in dilute - semidilute solutions in solvents selective for either the outer S block (dioxane) or for the middle EB block (heptane or decane or mineral oil). Equilibrium structure factors were measured over the temperature range of 20 - 180 C for different concentrations of the copolymer, ranging from 4% -20% (w/v). Our results showed that micelles were formed in both types of selective solvents. In the case of dioxane the micelles are isolated whereas in the case of heptane or decane or mineral oil a bridged micellar structure may be formed at higher copolymer concentrations. The core-shell structure of the micelles was examined using contrast matching SANS measurements for SEBS in heptane+deuterated heptane. In both heptane and dioxane we observed an ordered cubic structure of insoluble domains (micellar cores) at high concentrations (>8%). The micellar scattering function was fit to the Percus-Yevick interacting hard sphere model. In dilute micellar solutions an additional Lorentzian scattering can be identified, reflecting fluctuations. The temperature dependence of the core radius, the hard sphere interaction radius and the volume fraction of hard spheres was obtained. We have also obtained static structure factor data in pentablocks of styrene and butadiene, S-B-S-B-S in both heptane and dioxane by using SANS. We find that the triblock forms micelles at lower concentrations than the pentablocks.
We have used synchrotron-based time-resolved SAXS to examine the kinetics of (i) the nucleation and growth of micelles following a temperature jump from 90 C to 55 C in a 4% solution of SEBS in heptane, and (ii) ordering of the micelles on a cubic lattice for many different temperature jumps into the ordered cubic phase starting from the disordered micellar fluid phase in different solvents at different concentrations. Reversibility of these transitions has also been examined. The time evolution of the structural changes was determined by fitting the data with Gaussians to describe the structure factor of the ordered Bragg peaks and the Percus Yevic structure factor was used to describe the micellar fluid. The time dependence of the peak amplitudes and widths as well as of the micellar parameters will be presented. The results were analyzed using the Mehl Johnson Avrami theory of nucleation and show the kinetics of the transformation from the fluid to the ordered phase.