Special lectures: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17
W. GLEISSLE
Institut für Mechanische Verfahrenstechnik und Mechanik,
Universität Karlsruhe (TH), D-76128 Karlsruhe, Germany
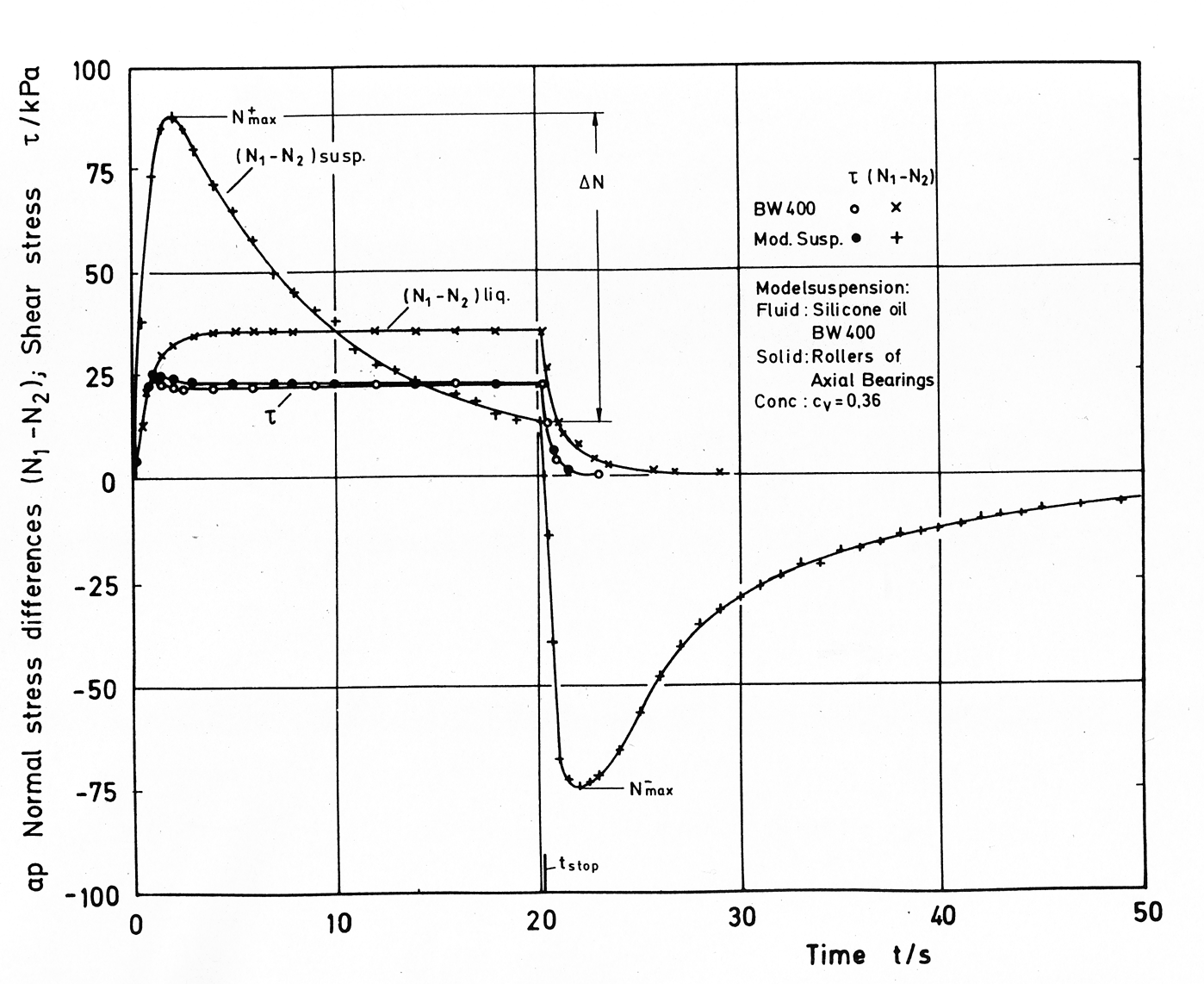
Measurements on concentrated suspensions, formulated with viscoelastic matrix liquids, indicate lower normal stress differences as in the clear matrix liquid. The first normal stress difference in a suspension decreases with increasing solid fraction if compared at constant shear stress. Simultaniously the transient behaviour of the normal stresses changes dramaticaly. Whilst the transient shear stresses of the matrix and the suspension behave similar, the normal stress growth at the begin of a shear experiment is accellerated in comparison to the normal stresses in the clear liquid. At high solid concentrations the growth of the normal stress is as rapid as the growth of the shear stress. Very high normal stress maxima and tremendous negative normal stresses can be observed after sudden cessation of the shear flow, Fig.1. It can be assumed, that the steep and rapid increase of the axial force (generated in a torsional rheometer), the high value of the normal stress maximum after start-up, its relatively slow decrease after passing the maximum and the high negative axial force are caused by different internal processes. One process-generating the positive axial force-likely has a short relaxation time, while another process-generating a negative axial force-likely has a long "relaxation" time. At steady state shear both processes are in equilibrium. Measurements of the pressure distribution in the shear gap of a parallel plate and a cone and plate rheometer were carried out, indicating that the second normal stress difference can be of the same magnitude as the first normal stress difference.
Fig.1: Shear and normal stresses in a clear silicone oil and in a model suspension
V.Kulichikhin, E.Plotnikova, G.Kulichikhin, and A.Subbotin
Institute of Petrochemical Synthesis, Russian Academy of Sciences
29 Leninsky Pr., Moscow 117912 Russia
For polymer blends containing approximately equal contents of isotropic polysulfone (PSF) and liquid crystal copolyester of PET and hydroxybenzoic acid (PES), having very great difference in viscosities (~three decimal orders) in homogeneous shear field (cone and plate operating unit) the characteristic S-shaped flow curves has been observed. At low shear rates and high total deformations (>100 units) the blend's viscosity obeys to additivity rule, i.e., it is located near the PSF flow curve. At reaching shear stress ~103 Pa the jump-like decrease of viscosity to PES's branch proceeds. For explanation of this effect the morphology of treated samples (solid discs) after definite prehistory was tested. It was shown that on low shear rate branch of flow curve a set of concentric rings was formed, and those preserve at high shear rates. Removing the PSF by selective solvent and IR analysis of selected skeleton parts have indicated on alternating phase separation of strongly interacted polymers, i.e., the neighbour rings are enriched either PSF, or PES. With increase of shear rate the depth of such separation increases. Theoretical approach was based on analysis of individual droplet of disperse phase behaviour at shear deformation. There exist two kinds of motion of droplets depending on their size: stretching of large ones and radial migration of small ones. The conditions were formulated for stretching the droplet along circular stream lines without breaking-up. The radial force is initiated by difference of normal stresses, but estimation of migration rate has shown very small efficiency of this process. Such a situation is typical for slow flows. At high shear rates the another process becomes valid, namely spurt effect which could be stimulated by formation of entanglements cracks in the matrix PSF (as near solid surfaces, as near interfaces), causing the bending of flow curve. At these circumstances the low viscous PES becomes responsible for blend's rheological properties.
Y. AOKI
Chemical Science Laboratories, Mitsubishi Chemical
Corporation, 1, Toho-cho, Yokkaichi, Mie 510-8530, Japan
Acrylonitrile-butadiene-styrene (ABS) polymers are one of the typical rubber-modified polymers and consist of a continuous poly(styrene-co-acrylonitrile) (SAN) in which the rubber phase (polybutadiene grafted with SAN) is dispersed in the form of spherical particles. ABS polymers are made by copolymerizing S and AN monomers in the presence of rubber. During the polymerization, some of the SAN become grafted to the rubber. These grafted SANs are believed to act as a dispersing agent.
In this lecture, I first summarize rheological features of ABS polymer melts and show that the shear storage modulus G˘ and loss modulus G˛ at low frequencies are influenced by the rubber particle agglomeration.1) Then I explain that the dispersion of rubber particles strongly depends on the composition2) and amount of grafted SAN.3) The mismatching of AN% between grafted and matrix SAN causes the agglomeration of rubber particles and the second plateau region appeared in the G˘ vs. frequency plots. Reducing the composition mismatch minimizes particle agglomeration and rubber particles are finely dispersed. Rubber particles with insufficient graft coverage tend to agglomerate owing to van der Waals attraction and depletion attraction causes agglomeration of particles at high grafting degree, as determined essentially by the interaction free energy between two grafted particles.4)
ABS samples having three different AN contents of grafted SAN were prepared, and dynamic viscoelastic properties and nonlinear relaxation modulus were measured for the samples at various rubber contents. Time sweep experiments were performed at 240 °C and at a frequency of 0.04 rad/s on the 6 % polybutadienne samples. An increase of the viscoelastic functions with time was observed. However, the G˘ and G˛ did not increase for a sample with matching of AN %. No agglomeration of rubber particles could be detected by transmission electron microscope. Dynamic viscoelastic and large deformation stress relaxation measurements were performed for the ABS having good dispersion of particles. Second plateau was not observed below 20 % of rubber content and was observed above 20 % for the dynamic measurements. Damping function of ABS polymers obtained from the stress relaxation depends on the rubber content. Increasing the rubber content, the damping function exhibits strong strain dependence. The relationships between the morphology and long-time relaxation are discussed.
References 1) Aoki, Y. J. Soc. Rheol. Jpn. 1979, 7, 20. 2) Aoki, Y. and Nakayama, K. J. Soc. Rheol. Jpn. 1981, 9, 39. 3) Aoki, Y Macromolecules 1987, 20, 2208. 4) Hasegawa, R., Aoki, Y. and Doi, M. Macromolecules 1996, 29, 6656.
J. MEWIS, I. VINCKIER, P. MOLDENAERS
Department of Chemical Engineering, Katholieke Universiteit Leuven,
de Croylaan 46, 3001 Leuven, Belgium
The relation between the rheological behaviour and the variable microstructure in polymer blends is investigated using uncompatibilized model systems. The linear viscoelastic properties are well described by the Palierne model. This model can be used to determine the size of the dispersed phase and its dependence on shear history. By using the Choi-Schowalter model or the more detailed description by Maffettone et al., morphological information can be derived from the steady state shear properties as well. Structural predictions for the various models are compared and the effect of shear rate on droplet size is discussed. At low volume fractions of the dispersed phase and low shear rates structural hysteresis phenomena can be detected. It is demonstrated how the presence of such a region can be exploited to study coalescence without interference of droplet break-up.
The transient rheological behaviour of blends is quite different from that of ordinary polymers, at least as far as the interfacial contribution is concerned. The latter is closely related to the evolution of the shape of the interface. Therefore suitable transient rheological experiments can provide detailed information about the flow-induced structural changes; inversely the transient rheological response can often be predicted quite well from the known evolution of the microstructure. Examples are given for start-up of flow, for a sudden increase or decrease of the shear rate, for flow reversal, for cessation of flow and for recoil. Droplet deformation, break-up and coalescence are considered. Starting from structural models, based on the Doi-Ohta approach, scaling relations can be derived for various transients.
In specially designed experiments, i.e. relaxation after an interrupted step-up in shear rate, droplet retraction and different mechanisms of break-up can be distinguished from the rheological response. The stress relaxation caused by a breaking thread can be analysed for dilute, Newtonian systems. Based on this analysis, a procedure has been designed to deduce the break-up time from the stress response. This approach is also valid in cases for which no theoretical analysis exists to date.
R. MULLERa*, B. ERNSTb, J.F. KOENIGb
aECPM, Polymer Processes and Materials, 25 rue Becquerel F-67200 Strasbourg, France
bELF-ATOCHEM, Cerdato-LEM, F-27470 Serquigny, France
The purpose of this work was to characterize by rheological measurements the interfacial grafting or crosslinking reaction in reactive polymer blends.
Due to their low melting temperatures, a series of ethylene-acrylate statistical terpolymers carrying either maleic anhydride or glycidyl methacrylate functional groups allowed to separate the formation of a controlled morphology from the chemical reaction at the interface : step 1 was carried out by steady shearing in a parallel plate rheometer at a temperature just above the melting temperature of the two components and for which the reaction betwen anhydride and epoxy groups was very slow. During step 2, the temperature was suddenly rised and the viscoelastic properties of the same sample were followed during the reaction by small amplitude dynamic measurements, which do not further affect the morphology. Several functional groups per chain were present for all polymers, which led to the formation of crosslinked interfacial layers.
The influence of various parameters could be investigated : the reactivity and miscibility of the blend components, the blend composition, the time and temperature of the reaction in step 2, the morphology at the end of step 1 depending of the amount of applied shear, the time allowed for diffusion of the reactive chains at the interface at the end of step 1.
It was also shown how the final blend properties are affected if both steps are carried out simultaneously at high temperature, i.e. if the rate of the interfacial reaction is of the same order than the rate at which the interface is generated by shearing.
The blends after reaction were characterized by solvent extraction, swelling and electron microscopy. The results were in agreement with those obtained from viscoelastic measurements.
* Present address : ELF-ATOCHEM, Cerdato-LEM, F-27470 Serquigny, France
M. GRMELA(1), C. HUMBERT(1),(2) and M. BOUSMINA(2)
Concentrated suspensions of spherical particles are investigated on three levels of description: the microscopic level, the level of kinetic theory, and the level of extended hydrodynamics. States of the suspended particles are characterized on these three levels by position and velocity vectors of the suspended particles, one particle distribution function, and hydrodynamics fields together with the Reynolds-like stress field respectively. The time evolution equations on all three levels are constructed as three realizations of one unifying Hamiltonian structure.
M. Bousmina, M. Grmela
Self and mutual diffusion at symmetric and non-symmetric polymers were studied by both attenuated total reflection infrared spectroscopy (ATR-FTIR) and rheometry. Both techniques showed that the mutual diffusion coefficients were significantly dependent on polymer molecular weight and molecular weight distribution of the medium of diffusion. For ATR-FTIR measurement a time-independent mutual diffusion coefficient was obtained, for which the experimentally determined concentration profile was fitted by using a combination of Fickian and Case II models. For rheometry technique a time-dependent mutual diffusion coefficient was obtained by transforming the evolution of G* with the time of welding to self and mutual diffusion coefficients using both reptation and fast theory. Previous models of Graessley and of Jabbari and Peppas were recovered as special cases of a general formulation for the diffusion process in polymer matrixes.
G. MARRUCCI, F. GRECO, G. IANNIRUBERTO
Dipartimento di Ingegneria Chimica, Universita Federico II, Piazzale Tecchio 80, 80125 Napoli, Italy
The theory of Doi and Edwards for entangled polymers has been recently modified for the case of fast flows to account for convective contributions to molecular dynamics [1,2]. The flow-induced relative motion between neighbouring chains removes constraints and speeds up relaxation. Convective constraint release (CCR) may thus explain why the shear stress is seen to approach a plateau at high shear rates instead of decreasing as predicted by the basic theory.
Returning to slow flows, another discrepancy between theory and observations can be found in the normal stress ratio in shear Y = - N2/N1. The theoretical value for Y is 1/7 whereas measured values are systematically larger, i.e., ca. 0.25 [3]. One way of predicting larger Y values while maintaining the classical tube picture is through elliptical tubes instead of circular ones, ellipticity being induced by deformation [4].
More generally, however, we suspect that discrepancies may arise because force balance requirements at the entanglement nodes are ignored in the classical theory. Since chains carry an essentially constant tension of order kT/a (where a is tube diameter), the force balance on a node couples local chain orientations. Consequences of this concept are being explored.
Montgomery T. Shaw,a,b Yong-Woo Inna and L. Wang,a Polymer Programa and Department of Chemical Engineering,b Institute of Materials Science, University of Connecticut, Storrs, CT USA
Sharkskin melt fracture (SSF) occurs at a shear stress of ~0.1 MPa, giving a matt surface on the extrudate. We examined time-resolved rheo-optical signals to find if SSF develops within the die land. We also searched for a discontinuity in the slope of the flow curve. [Inn et al. (1998)] We chose polybutadiene (PBD, Mw = 182,000 and Mw/Mn = 1.8) because it exhibits large SSF and has a high stress-optical coefficient. Rod dies (12.7 mm w/ L/D = 4, 6, 10) were attached to a 19-mm single-screw extruder operated at 50 oC. Rheo-optical studies used a 2x25-mm slit die. Measured were 2d LALS, time-resolved Hv and Vv scattering at fixed angles, time-resolved birefringence and particle velocities.
Below are typical particle-tracking results and the transform to the frequency domain.
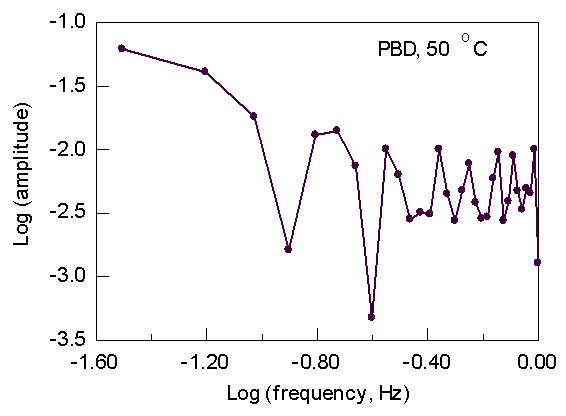
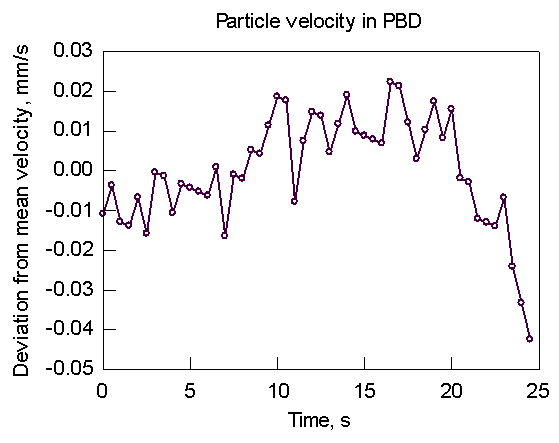 Fig. 1. Particle
velocity Fig. 2. Transform of Fig. 1
Fig. 1. Particle
velocity Fig. 2. Transform of Fig. 1
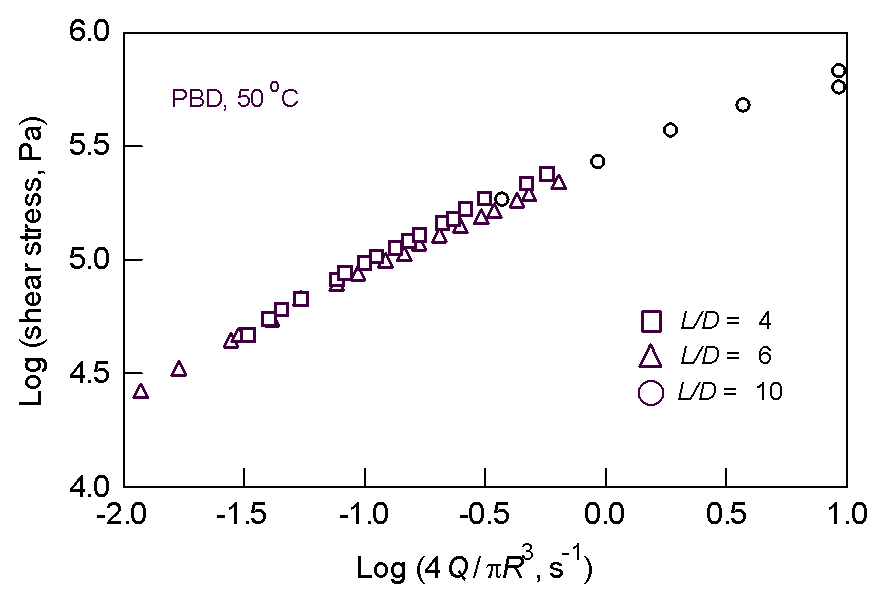
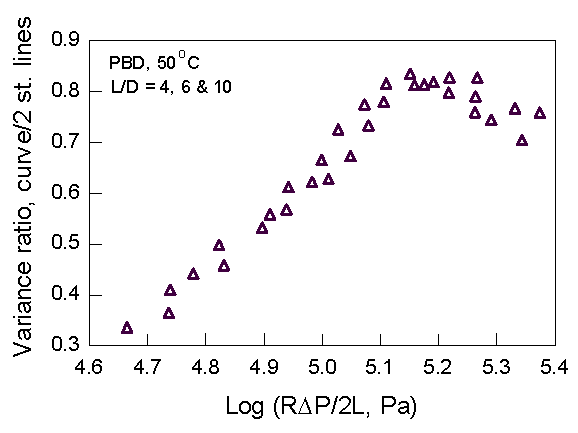 Fig. 3. Flow curve
for PBD Fig 4. Breakpoint analysis of Fig. 3
Fig. 3. Flow curve
for PBD Fig 4. Breakpoint analysis of Fig. 3
Clearly, there is no sign of any periodic disturbance in the velocity near the SSF frequency of 0.2 Hz. (-0.7 on the log scale). A statistical analysis of the PBD flow curve (Fig. 3) provides a slight indication of a "kink" at around 5.2 log stress (Fig. 4). However, this stress is about a factor of 2 higher than the observed stress at which SSF starts. The general conclusion is that if SSF starts in the die land, it is very subtle indeed.
REFERENCE: Y-W. Inn, R. J. Fischer, M. T. Shaw, Rheol. Acta 37, 573-582 (1998)
M. D. HAW and P. NAVARD
Ecole des Mines de Paris, Centre de Mise en Forme des Matériaux (CEMEF), UMR 7635, B.P. 207, 06904 Sophia Antipolis, France
The flow of a "model" lyotropic liquid crystal polymer, (hydroxypropyl)cellulose in water, through a rectangular channel with a divergence in the channel width, is studied by in-situ light microscopy.
Microscopic texture observations are related to measurements of the flow velocity field, in order to characterize the shear and elongational aspects of the flow and examine the effects of the divergence from the narrow entry channel to the wide channel. A strong dependence of flow-induced texture on position in the channel is observed and is related to the interplay of shear and elongational strain. The divergence generates both a perpendicular elongational strain due to the widening of the channel, and subsequently an elongational strain along the flow direction due to the change in flow pattern from quasi-radial to unidirectional along the wide channel. Additionally side wall structure is observed to be more complex than a simple strong alignment, displaying a fine birefringent texture. Finally there is a marked dependence of the macroscopic structure on the strain history of the fluid prior to entry into the channel, indicating that very different structures of, for instance, moulded parts, can result from differences in geometry and fluid treatment prior to entry into the mould itself.
This work has been sponsored by the European Community through the Training and Mobility of Researchers programme (TMR contract 96-0003)
OF COMPLEX MACROMOLECULES
T. PAKULA
Max-Planck-Institute for Polymer Research, Postfach 3148, 55021 Mainz, Germany
Various macromolecular objects, such as multiarm stars, hyper-branched and dendritic polymers, microgels, or micelles show in melts well developed structures which result from strong excluded volume interactions between the individual relatively compact macromolecular elements. This ordering involves extra slow relaxation processes which determine the terminal flow behavior of melts. Results concerning structure and rheology of such melts will be presented and discussed. The small angle X-ray scattering as well as the mechanical and dielectric spectroscopy methods are used.
Experimental results are compared with simulation of structure and dynamics in corresponding model macromolecular systems. Simulations have been performed using the Cooperative Motion Algorithm. Simulated model melts of multiarm stars and microgels indicated qualitatively the same effects as observed in real systems i.e. the ordering of macromolecules and the corresponding slow relaxation modes.
Based on a detailed observation of the dynamics in such systems, a mechanism for the structural relaxation is suggested which consists in cooperative rearrangements involving synchronized displacements of a number of neighboring macromolecules which escape in this way from the "cages" formed by the almost non penetrable macromolecular neighbors.
Fig.1. Illustration of
some macromolecules
showing ordering and
structural relaxation
modes in melts.
stars microgels
B. VERGNES, F. BERZIN
CEMEF, Ecole des Mines de Paris, UMR CNRS 7635,
BP 207, 06904 Sophia-Antipolis Cedex (France)
The control of the molecular weight distribution of poly(propylene) resins by peroxide degradation is widely used in polymer industry. It allows to adjust the viscosity of these resins to the level required for processing applications. The purpose of this work was to characterise the influence of peroxide degradation on rheological behaviour of both homo- and copolymers, and to use these results to obtain a predictive model of the degradation in a twin-screw extrusion process.
The polymers studied were an homopolymer (Appryl 3050BN1) and a block copolymer (Appryl 3060MN5). Peroxide was a DHBP (2.5-dimethyl-2.5-di(t- butylperoxy)hexane ) Trigonox 101 (Akzo Chemie). Samples were prepared on a co-rotating twin screw extruder (Leistritz 30-34) with various amounts of peroxide (0.01 to 0.5 wt. %), and characterised by gel permeation chromatography and rheological measurements in low amplitude oscillatory shear. As expected, for both polymers, the average molecular weight and the width of the distribution decreased when increasing the amount of peroxide. However, a tail of high molecular species was observed for the copolymer, even at high peroxide concentration. The rheological characterisation confirmed the classical behaviour of the homopolymer. Its viscosity decreased and became more Newtonian when peroxide concentration increased. For the copolymer, results were different : an increase of viscosity was observed at low frequencies, which can reach more than one decade for the more degraded products. This behaviour is due to the presence of the ethylene phase, leading to a "blend" with a high viscous phase dispersed in a low viscous Newtonian matrix.
Degradation experiments were carried out on a co-rotating twin screw extruder (Werner & Pfleiderer 30), in various conditions of feed rate, screw speed, barrel temperature and peroxide content. Samples were collected at the die exit and analysed. Coupling a thermomechanical model of the twin screw extrusion process [1], a kinetic model of the considered reactions [2] and the rheological behaviour previously described, it was possible to calculate the change in molecular weight along the extruder, during the peroxide-controlled degradation. The peroxide efficiency was supposed to be only dependant on the peroxide concentration. The results of the computation are in good agreement with the measurements made at the die exit.
[1] B. Vergnes, G. Della Valle, L. Delamare, Polym. Eng. Sci., 38, 1781 (1998)
[2] M.J. Krell, A. Brandolin, E.M. Valles, Polym. React. Eng., 2, 398 (1994)
G. EDER
Institute of Chemistry, Linz University, Altenbergerstr. 69, A-4040 Linz, Austria
The modeling of the solidification behavior of semi-crystalline polymers during processing is quite a complex task. In general one has to do with a flowing and simultaneously solidifying melt. Up to now there are practically no studies about the rheological properties of solidifying melts, at least in the interesting range of deformation conditions as occurring in processing. On the other hand one has also a dramatic change of the morphology of processed polymers as compared with the same material crystallized under quiescent conditions. Usually the morphology is made responsible for the properties of a solidified product. Thus a thorough understanding of the kinetics of crystallization under flow conditions is of eminent importance for the production of high-quality plastic parts.
The main difficulty in the analysis of structure formation in practical processing lies in the fact, that the crystallization kinetics is heavily dependent on temperature (which is also true in the much simpler case of crystallization in quiescent melts) and on the deformation history of the melt. Due to the latent heat evolving during crystallization the temperature history, flow history and the course of the crystallization process are strongly coupled. The only method to get insight into this complex problem is the use of carefully selected model experiments keeping most of the relevant physical parameters constant, and varying some others in a range as relevant in polymer processing (e.g. high shear rates).
In a series of experiments, which were chosen in such a way that the time period of deformation and the time period of crystallization do not overlap, a few important results could be obtained, which makes it possible to derive sound theoretical models for the crystallization kinetics under deformation conditions:
G.KERCH
Centre of Science and Engineering EKOTRA, Nicgales 4 - 235, LV- 1035, Riga, Latvia
A number of phenomena, characterized by drastic changes in mechanical and physical properties during the flow of polymer melts and solutions or during the mechanical deformation of polymer solids can be considered from a single point of view. The process of high-speed melt spinning, flow induced phase separation in polymer solutions, critical coil-stretch transition for flexible molecules, abrupt neck-like deformation of polymers, abrupt shear thickening of the viscosity in the vicinity of critical regions, negative thyxotropy, sharp increase of effective surface energy, stress intensity factor, dynamic fracture toughness during the dynamical fracture of solid polymers are the examples of such phenomena. These changes in properties may be associated with energy and inertia increase when velocity of extension for polymer solids and melts or shear rate for polymer solutions approaches the certain limiting value.
It is possible to predict, at least qualitatively, the flow induced changes in temperatures of phase separation in polymer solutions if we shall take the free energy of mixing n1 moles of solvent with n2 moles of polymer to be given by modified Flory-Huggins equation
D GM = RT[n1ln(1 - j ) + n2 lnj +c 1(1 - j )N]/(1 - v2/c2)1/2
where R is the gas constant, T is absolute temperature, j is the volume fraction of polymer, and N is given by N = n1 + m n2 , with m the molar volume of polymer divided by the molar volume of solvent; c 1 is the interaction parameter, v is velocity of flow, c is the limiting velocity.
The manifestation of relativistic effects in polymer solutions, melts and solids and instabilities in the vicinity of critical regions are discussed. The realization of the significance of the limiting velocity and sharp increase of energy and inertia in the neighborhood of limiting velocity gives the opportunity to develop a new approach to the consideration of rheological behaviour of polymers.
I. FORTELNÝ
Institute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, Heyrovského nám. 2, 162 06 Praha 6, Czech Republic
An adequate model of the coalescence is necessary for correct microrheological description of the phase structure development in immiscible blends at rest and in flow.
The coalescence in quiescent molten polymer blends is caused by molecular forces and Brownian motion. The coalescence rate is controlled by drainage of the matrix film between undeformed droplets. The theory of coalescence induced by molecular forces or Brownian motion predicts the rate of coalescence in qualitative agreement with experimental data. The coalescence is faster in a viscoelastic than in the Newtonian matrix with the same viscosity. Description of the coalescence induced by the Brownian motion should be improved and mutual influence of the Brownian motion and molecular forces should be considered. Simultaneous interactions of three and more droplets, which can have a fundamental effect on the dependence of the coalescence rate on the blend composition, should be investigated.
Probability, PC, that the collision of droplets in a shear flow is followed by their fusion, is controlled by the competition between their approach and rotation around their common centre of inertia. The force inducing approach of the droplets changes during their rotation. The velocity of the droplets approach is controlled by drainage of the matrix between flattened droplets. The results of the calculation of PC by Elmendorp's and Janssen's methods have been compared with the results for constant flattening and instantaneous adaptation of the droplet shape to the force during the rotation. The dependence of PC on properties of the components is determined by the model of interface mobility, the magnitude of PC depends on the description of the competition between approach and rotation of the droplets. The effect of variable driving force and viscoelasticity of the components on the droplet shape should be studied more in depth. The main shortcoming of the present theories of blends with high contents of the dispersed phase is namely neglecting any effect of other droplets on the collision of a certain pair of droplets.
Acknowledgement: The author thanks Dr. A. Živný for numerical calculations and Grant Agency of the Czech Republic for support by grant No. 106/99/0555.
MATTHIAS SCHNELL AND BERNHARD A.WOLF
Institut für Physikalische Chemie and
Materialforschungszentrum der
Johannes Gutenberg-Universität, Jakob-Welder-Weg 13, D-55099
Mainz, Germany
Email: Bernhard.Wolf@Uni-Mainz.de)
The viscosity of polymer/solvent systems is modeled theoretically as a function of composition under the premises that the dissipation of energy is taking place at the molecular interfaces and that the friction between solvent and solute varies with composition due to a change in the flow mechanism (drainage of coils). The simple expression obtained in this manner contains three system-specific parameters: A geometric factor g , which accounts for the differences of the surface to volume ratios of the components, a hydrodynamic parameter a , which measures the friction between solute and solvent in case of fully draining polymer coils, and b , which corrects for changes in the friction between unlike molecules resulting from collective motions owing to limited draining. Experimental data published for twelve poly(dimethylsiloxane)/pentamer mixtures can be represented quantitatively by this relation; moreover, the knowledge of the three system specific parameters permits the calculation of intrinsic viscosities, and the molecular weight dependencies of g and a yield the entangle molecular weight of the polymer.
VERENA ZIEGLER AND B.A.WOLF
Institut für Physikalische Chemie and
Materialforschungszentrum der
Johannes Gutenberg-Universität, Jakob-Welder-Weg 13, D-55099
Mainz, Germany
(Email: Bernhard.Wolf@Uni-Mainz.de)
Stationary state viscosities h were measured at 50 °C for two-phase blends consisting of poly(dimethylsiloxane) [PDMS] and poly(dimethylsiloxane-ran-methylphenylsiloxane) [COP] at different compositions as a function of shear rate up to 100 s-1. All mixtures exhibit shear-thinning behavior in contrast to the pure components; the sensitivity of h towards shear varies with composition in a characteristic manner for which we suppose that it reflects the steady state morphology of the blends. On the basis of these rheological results we postulate that a composition range of co-continuity should replace the concept of a single composition at which phase inversion happens. For blends consisting of droplets of one phase, suspended in the matrix of the complementary phase, pictures were taken of after rapid transfer from the shear cell (100 s-1) into the light microscope. The average dimensions of the droplets are in both cases found to be approximately 13 mm regardless whether the matrix consists of PDMS or COP; this observation is in contrast with calculations on the basis of the interfacial tensions and viscosity ratios of the two phases, which we have measured as a function of temperature. For the PDMS matrix this prediction (24 mm) is reasonable; however, if the matrix consists of COP, the theoretical result exceeds the measured dimension by approximately a factor of 14.