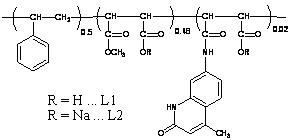Krátká sdělení
1 2
3 4
5 6
7 8
9 10
11 12
13 14
15 16
17 18
19 20
21 22
23 24
25 26
27 28
29
MOŽNOSTI APLIKÁCIE SLEDOVANIA
SLABEJ SVETELNEJ EMISIE DOPREVÁDZAJÚCEJ OXIDÁCIU POLYMÉROV
J. RYCHLÝ, L. RYCHLÁ
Ústav polymérov SAV, Centrum excelentnosti CEDEBIPO, Dúbravská
cesta 9, 842 36 Bratislava
Slabá sveteľná emisia (chemiluminiscencia), ktorá doprevádza
oxidáciu polymérov, má niekoľko aspektov praktického využitia. Ide
najmä o možnosť stanovenia indukčnej periódy oxidácie v prípade
termickej oxidácie stabilizovaných polyolefínov a polydiénov, ktoré je
ekvivalentné podobným stanoveniam napr. z absorbcie kyslíka alebo zo
zmeny mechanických vlastností. Vysoká citlivosť photon-counting systému
umožňuje rozlišovať rozdiely v rýchlosti oxidácie už na začiatku
sledovaného procesu a na teplotnej škále je možné sa posunúť prakticky
do oblasti laboratórnych teplôt. Neizotermické postupy s programovaných
ohrevom alebo ochladzovaním urýchľujú posúdenie oxidovateľnosti
akéhokoľvek organického materiálu s možnosťou viac alebo menej
spolahlivej aproximácie k laboratórnym podmienkam. Hydroperoxidy v
polymérnych materiáloch takisto vyžarujú chemiluminiscenciu pri svojom
rozklade, čo umožňuje stanovovať ich zvyškový obsah. V posledných
rokoch metóda našla nové uplatnenie pri hodnotení zvyškovej stability
historických dokumentov na báze papiera, pri štúdiu stability
muzeálnych a iných artefaktov na báze polymérov a najmä pri opise
degradovateľnosti biopolymérov, ako je škrob, celulóza, dextrán,
pullulán a i. Rozsah informácií o oxidovanom systéme, ktorý je možný
získať, bude v prezentovanej práci doplnený experimentami pri rozličnom
zložení oxidujúcej atmosféry (Obr. 1), relaxačným postupmi náhlej zmeny
zloženia oxidujúcej atmosféry alebo skokovitej zmeny teploty. Keďže ide
o metodiku, u ktorej emitované svetlo môže tienené svetelnými
absorbérmi, zhášačmi excitovaných stavov alebo inými svetlnými
filtrami, je potrebné, aby chemiluminiscenčné experimenty boli
doprevádzané aj výsledkami iných metód. Pre rozličné polyméry budú
uvedené porovnania s metódou DSC, termogravimetrie, absorbcie kyslíka a
spektrálnymi metódami stanovenia karbonylov.
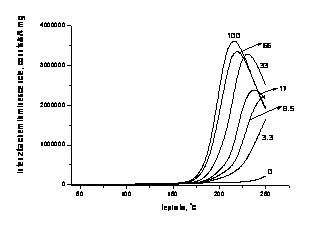
Obr. 1. Neizotermické priebehy intensity chemiluminiscencie
pri
oxidácii poly (vinyl pyrolidónu) pri rozličných koncentráciách kyslika
v oxidujúcej atmosphere. Rýchlosť ohrevu 2.5 oC/min
v rozsahu teplôt 40 - 250 oC
STUDY OF PHOTOPEROXIDATION AND CROSSLINKING OF STYRENE
COPOLYMER
BEARING BENZIL PENDANT GROUPS USING FLUORESCENCE PROBES AND
CHEMILUMINESCENCE
C. KÓSAa, T. CORRALESb,
C. PEINADOb , M.J.GARCIA-CASASb,
I.LUKÁČa,
a Polymer Institute, Slovak Academy of
Sciences, Dúbravská cesta 9, 842 36 Bratislava, Slovak Republic,
(upolkosa(savba.sk)
b Departamento de Fotoquímica de
Polímeros Instituto de
Ciencia y Tecnología de Polímeros, C.S.I.C. Juan de la Cierva 3,
28006-Madrid, SPAIN.
BZMA/S copolymer films were prepared and irradiated at l
>400 nm in the presence of oxigen, such as the
phototransformation
of benzyl to benzoyl peroxide pendant groups takes place, and their
thermal decomposition produced the polymer crosslinking.1
In this work, 2 commercial stilbene like D- p- A+X-
type fluorescent probes containing pyridinium (PYRIDINE 1) and
benzthiazolium groups (STYRYL 7) and an intramolecular excimer forming
fluorescent probe, DiPyM (Scheme 1), has been chosed as a valuable
method to analyse the crosslinking process. While the stilbene like
probes, STYRYL 7 and PYRIDINE 1, the changees of emission maxima, 2
in the case of DiPyM the rate of excimer emission increase during its
outdiffusion from the crosslinked polymer was utilized to observe the
information about the polymer network density.3
Comparing the UV spectra of benzil groups and the UV spectra
of
PYRIDINE 1 and SYRYL 7 show, that these probes are can not be used to
followe the crosslinking during benzil group photooxidation due to
possible reaction with excited benzil groups. On the other hand,
photophysical properties of DiPyM shows possible utilization (not
absorb at the irradiation wavelength).
In general, the fluorescence intensity of DiPyM, at shortest
wavelength which corresponded to monomer emission of probe, increased
during irradiation of the samples. By excitation the sample at 345 nm
beside the DiPyM molecule, the benzil moieties are excited too.
Therefore, the increase of DiPyM monomeric fluorecence intensity is due
to increase of the light absorbed by DiPyM molecule upon consumption of
benzil moieties. Increase of DiPyM excimer emission by time was
observed upon immerzion of irradiated and iradiated and thermally
treated BZMA/S copolymer film to cyclohexane solution.
A good correlation observed between fluorescence, FTIR and CL
measurements during photochemical formation and thermal decomposition
of peroxides has been found. The chemiluminescence emission was seen to
enhance with irradiation time, which would be related to the benzoyl
peroxide moieties generated during irradiation. The increase of
chemiluminescence intensity was interrupted at longer time of
irradiation, when concentration of those species tended to a nearly
constant value, as it was observed by FTIR. In this case, others
factors may be considered to affect the chemiluminescence emission, for
example the increasing crosslinking on irradiated samples, which would
restrict the mobility of hydroperoxides to react due to the
crosslinking of the network.
The results obtained contributes to the development of a sensitive
fluorescence based method in order to assess photocrosslinking of a
material in the early stage of the process, due to its sensitivity
comparable to that of chemiluminescence analysis.
| Scheme1 |
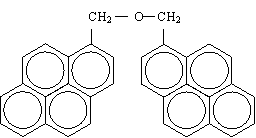 |
|
| DiPyM |
|
References:
1. Kósa Cs., Lukáč I., Weiss R. G. : Macromol. 33,
4015 (2000)
2. Peinado C., Salvador EF., Baselga J., Catalina F.:
Macromol. Chem. Phys., 202, 1924 (2001)
3. Danko M. Hrdlovič P. Borsig. E.: Polymer 44,
389 (2003)
UTILIZATION OF FLUORESCENCE SPECTROSCOPY FOR LIVING RADICAL
POLYMERISATION OF STYRENE
Ľ. BÚCSIOVÁa , M. YINb,
W.D. HABICHERb
a Polymer Institute SAS, Bratislava,
Slovak Republic
b Institute of Organic Chemistry, TU
Dresden, Dresden, Germany
The field of living free radical
polymerisation1
has rapidly expanded in the recent years. Nitroxide-mediated technique
of living polymerisation is usually used for styrene-based systems, and
it has significant functional group tolerance and easy purification of
obtained polymer2.
The synthesis of functionalized unimolecular initiators3
permits the preparation of wide range of different materials which are
either difficult to prepare or not available via other polymerisation
processes.
In present work is described the preparation of new
unimolecular
initiator with photosensitive properties and their usage for
polymerisation of styrene. New unimolecular initiator with
photosensitive properties 2,2,6,6-tetramethyl-1-(1-phenylethoxy)piperidin-4-yl
4-pyren-1-ylbutanoate
(KO) was prepared. UV and emission spectra were measured and evaluated
in comparison with nitroxide radical covalently bonded with chromophore
{2,2,6,6-tetramethyl-4-[(4-pyren-1-ylbutanoyl)oxy]piperidin-1-yl}oxidanyl
(PYNO) in methanol, cyclohexane and toluene. Polymerisation of styrene
with new unimolecular initiator was typical nitroxide mediated living
radical polymerisation. For comparison, two different ratios (1:400 and
1:1000 initiator - monomer [I:M]) were used for polymerisation. When
[I:M]=[1:400], the obtained polydispersity was 1.14-1.16 and molecular
weight 29900 - 30850 g/mol. For ratio [I:M]=[1:1000], the
polydispersity was 1.25-1.26 and the molecular weight 66550 - 68760
g/mol. The conversion was for [1:400] 62 - 65% and for [1:1000] 76 -
79%. The time dependence of the conversion was in both cases
unimolecular and could be described with a kinetic equation of first
order. The fluorescence intensity was followed during the
polymerisations and the data acquired with initiator with fluorescening
group could clearly demonstrate the living manner of polymerisation.
For comparison, two different ratios
initiator to
monomer [I:M] were used ([I:M]=1:400 and [I:M]=1:1000). At higher
concentration of monomer [I:M]=1:400, the final molecular weight was
lower and reached more rapidly, due to the higher amount of reaction
centre, and the polydispersity narrower was in this case . The time
dependence of fluorescence intensity on polymer during polymerisation
was measured. The fluorescence intensity of all polymer samples, in
both of initiator/monomer ratio, was less intensive than the intensity
of pure initiator. During the polymerisation the emission intensity was
decreasing, what was surprising, but we attempted to give an
explanation for this effect.
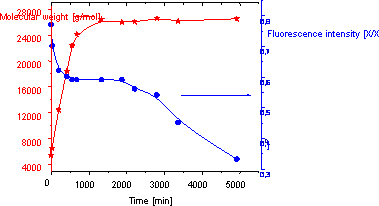
Fig. 1 Time dependence of
molecular weight [x] and fluorescence intensity of polymer samples [,]
(ratio [I:M]=[1:400])
References
1Fischer,
H. Macromolecules 1997, 30,
5666; Hawker, C. J. Acc. Chem. Res. 1997, 30,
373; Colombani, D. Prog. Polym. Sci. 1997, 22,
1649; Matyjaszewski, K. Controlled Radical Polymerisation;
ACS Symposium Series 685; American Chemical Society: Washington, DC,
1998.
2
Hawker,C. J.; Bosma, A. W., Hart, E. Chem. Rev.
2001, 101, 3661
3
Hawker, C. J.; Barclay, G. G.; Dao J. J. Am. Chem. Soc.
1996, 118, 11467; Li, I. Q.; Howell, B. A.; Koster,
R. A.; Priddy, D. B. Macromolecules 1996, 29,
8554; Connolly, T. J.; Baldov, M. V.; Mohtat, N.; Scaiano, J. C.Tetrahedron
Lett. 1996, 37, 4919; Bergbreiter, D. E.;
Walchuk, B.Macromolecules 1998, 31,
6380; Woodworth, B. E.; Zhang, X.; Gaynor, S. G.; Metzner, Z. Macromolecules
1998, 31, 5955; Wang, D.; Wu, Z. Macromolecules
1998, 31, 6727; Gravert, D. J.; Janda, K. D. Tetrahedron
Lett. 1998, 39, 1513.
LANTHANIDE(III) ION LUMINESCENCE IN COMPLEXES WITH POLYMER
LIGANDS
S. KUKLA, D. VÝPRACHTICKÝ, V. CIMROVÁ
Ústav makromolekulární chemie, Akademie věd České Republiky,
Heyrovského nám. 2, 162 06 Praha 6, Česká Republika
kukla(imc.cas.cz, vyprach(imc.cas.cz
Introduction
The importance of lanthanide(III) ions luminescence stems from
its
peculiar characteristics. Terbium(III) ion which I used in most of the
experiments exhibits a long-lived (up to milliseconds) excited state 5D4
and its emission spectrum consists of several sharp bands. This is due
to the fact that the 5D4
emitting excited state and all the 7FJ
(J = 0, 1, …, 6) terms of the ground state multiplet
have the same 4f8 electronic
configuration and that the 4f electrons are well
shielded from external charge by outer 5s2
and 5p6 shells. Electric
dipolar transitions which involve only redistribution of electrons
within the 4f
orbitals are strictly parity-forbidden by the Laporte rule. In
addition, a number of the discussed transitions are also forbidden by
the spin cross-over rule. Concerning potential applications of
lanthanide ions luminescence, the parity-forbidden nature of the f-f
transitions is the major draw-back. These transitions have very low
molar absorption coefficients (of the order of unity) and, thus, the
direct excitation of the emitting metal ion is inefficient. Moreover,
the desirable intrinsic luminescence can be quenched by nonradiative
dissipation of excitation energy into suitable vibrational modes of the
environment, the process becoming more probable when vibrational quanta
of the oscillators or their overtones match the energy gap between the 5D4
emissive level and the highest 7F0
level of the ground state multiplet. When solvent molecules containing
O-H groups (such as water or methanol) are coordinated to terbium(III)
ion, this deactivation pathway becomes very efficient. On the
assumption that every O-H oscillator quenches independently, a
quantitative method for estimation of the number of solvent molecules
in the first coordination sphere of terbium(III) ion was proposed. It
is based on a deuterium isotope effect: if the O-H oscillators (3500 cm-1)
are replaced by the low-frequency O-D oscillators (2800 cm-1),
the vibronic deexcitation pathway becomes much less efficient owing to
a smaller Franck-Condon overlap between the two vibronic wavefunctions
and, consequently, the observed luminescence lifetime of terbium
increases. This concept makes terbium luminescence a valuable
tool
for obtaining information on the most intimate microenvironment of the
ion - the polymer micro-domain in our case.
Design of polymer ligands
The design of the polymer ligand was based on the general
knowledge that it should:
-
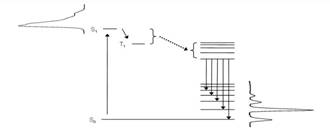 |
| Figure
1 |
contain a suitable, strongly absorbing (organic)
fluorophore for an efficient energy transfer to the otherwise weakly
absorbing Tb3+ (Jablonski diagram for this
process which is usually referred to as "antenna effect"
is plotted in Figure 1);
- be able to tightly bind the ions. As it is assumed, the
macromolecular nature of the ligand then ensures shielding of the bound
ions and prevents deactivation of their excited states by the solvent
environment. According to Pearson's theory, lanthanide(III) ions are
hard acids. Much of their coordination chemistry involves anionic
oxygen donors and it is well established that carboxylates
coordinate the lanthanide(III) ions very well.
Using the methods of polymer chemistry, these requirements
were matched by this synthetic strategy: polymeranalogicalreaction
of parent poly[styrene-alt-(maleic anhydride)] with
7-amino-4-methylquinolin-2(1H)-one (energy donor)
and methanol, yielding ligand L1 and by its subsequent neutralization
and dialysis ligand L2 (see Figure 2)
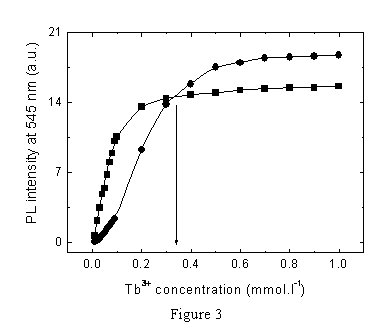
Luminescence studies
When excited at 345 nm, both L1 and L2
ligands show a
short-lived broad-band emission spectrum with maxima at 372 and 395 nm,
which are due to the bonded quinolinone fluorophore. When Tb3+
was added to ligands and a complex in methanol solution was formed, the
ligand emission intensity decreased. This was accompanied by an
increase in the intensity of the typical long-lived terbium(III) ion
emission bands at 490, 545, 585 and 620 nm, corresponding to the 5D4→7F6,
5D4→7F5,
5D4→7F4
and 5D4→7F3
transitions, respectively (see inset in Figure 3). The dependence of
terbium luminescence intensity of [Tb(III)-L1]
(Figure 3, squares) and [Tb(III)-L2] (Figure 3,
circles) on Tb3+ concentration was described in
terms of the donor-acceptor distance, employing the concept of "effective
concentration of fluorophore and binding sites inside the coil".
A qualitative model based on the phenomenon of adsorption (for
noncharged ligand bearing COOH groups) and chain expansion (for charged
ligand bearing COONa groups) was put forward. This concept was strongly
supported by time-resolved measurements in common and deuterated
methanol. The time-resolved luminescence revealed that the decay curves
were double-exponential with predominating longer component (rel B1
> 85 %) for both [Tb(III)-L1] and [Tb(III)-L2]
complexes. About 2.5 methanol molecules were coordinated to Tb3+
in [Tb(III)-L1] complex, while free Tb3+
coordinates approximately 6.2 ± 0.5 methanol molecules according to our
measurements. After neutralization the binding properties of the
polymer coil improved and, on average, two methanol molecules were
coordinated to terbium(III) ion in the [Tb(III)-L2]
complex. In addition, the number of coordinated methanol molecules
varies with Tb3+
concentration in this case. This is a consequence of the conformational
changes of the polyelectrolyte-type ligand, which was also verified by
viscosity measurements.
Steady-state measurements were also used to prove the
importance of
steric properties of macromolecular architecture in binding and in the
ligand-to-metal energy transfer process. When a model compound, N-(4-methyl-2-oxo-1,2-dihydroquinolin-7-yl)succinamic
acid, was added to a methanol solution of TbCl3,
no significant increase in terbium luminescence was detected. This
showed that a complex with Tb3+
in methanol solution was not formed, even though the acid bears the
same binding site and fluorophore unit as the synthesized ligands L1
and L2. On the other hand, we estimated that the
relative terbium luminescence intensity in complexes with the L1
and L2 ligands is approximately four orders of
magnitude higher that that for TbCl3 itself.
The described behavior of macromolecular ligands is attributed to the
"polymer cooperative effect", i.e. the high local concentration of
binding sites and fluorophores in the polymer domain ("effective
concentration inside the coil").
Acknowledgements
We thank the Grant Agency of the Czech
Republic for
support of this work (grant 203/04/1372) and the Grant Agency of the
Academy of Sciences of the Czech Republic (grant IAA4050409).
NOVÉ PRÍSTUPY V SYNTÉZE FOTOREAKTÍVNYCH POLYMÉROV
NA BÁZE
2-OXAZOLÍNOV
J. KRONEK*, J. LUSTOŇ
Ústav polymérov, Slovenská akadémia vied, Dúbravská cesta 9,
842 36 Bratislava, Slovenská republika, upolkron(savba.sk
Polyméry obsahujúce chromofór majú široké využitie v
polymérnej
chémii a technológii. Využívajú sa ako farbivá, fluorescenčné značky,
molekulové prepínače, fotodiódy, fotorezisty a pod. Jednou z hlavných
charakteristík chromofórov je, že obsahujú systém nenasýtených väzieb s
predĺženou konjugáciou, pričom sa môžu využívať násobné väzby C=C alebo
násobné väzby medzi heteroatómami (C=N, C=O, N=N, N=O).
Na zavedenie chromofóru do polymérneho reťazca je možné použiť
množstvo metód zahŕňajúcich prípravu monoméru obsahujúceho chromofór
alebo modifikáciu bežného polyméru. Kombináciou techník možno pripraviť
polymér obsahujúci chromofór v hlavnom reťazci, v bočnom reťazci alebo
na koncoch molekuly.
Jednou zo skupín zlúčenín vhodných na prípravu fotoreaktívnych
polymérov s rôznou topológiou sú heterocyklické zlúčeniny obsahujúce
2-oxazolínovú skupinu. 2-Oxazolíny, alebo tiež 4,5-dihydrooxazoly,
patria medzi reaktívne zlúčeniny so širokým využitím v organickej
syntéze [1], ako aj v chémii polymérov [2]. Poskytujú adičné reakcie s
karboxylovými kyselinami, tiolmi, fenolmi, amínmi alebo epoxidmi [1],
ktoré tiež možno využiť ako rastové reakcie v adičných polymerizáciách.
Ďalšou polymerizačnou reakciou 2-oxazolínov je katiónová polymerizácia,
ktorá patrí medzi kontrolované procesy a možno ju využiť na prípravu
polymérov s nastavením štruktúry [3].
Uvedené reakcie 2-oxazolínov boli použité na prípravu
fotoreaktívnych polymérov s rôznou topológiou. Ako chromofóry sa
použili multifunkčné zlúčeniny na báze 2-oxazolínov obsahujúce
nenasýtenú väzbu C=C, N=N ako aj kondenzované aromatické jadrá.
Vhodná architektúra sa nastavila využitím polyadičnej reakcie,
kde
sa pripravili polyméry obsahujúce chromofór v hlavnom reťazci [4],
katiónovej polymeriácie, pomocou ktorej sa chromofór zaviedol do
bočného reťazca polyméru, alebo modikačnej reakcie bežných polymérov
obsahujúcich vhodnú reaktívnu skupinu (polyetylénglykoltereftalát,
kyselina polyakrylová) [5,6]. Fotochemické vlastnosti sa sledovali
UV/Vis spektroskopiou alebo fluorescenčnou spektroskopiou, pričom sa
sledovala závislosť zmeny vlastností od štruktúry polymérov.
Autori ďakujú Slovenskej grantovej agentúre
za finančnú
podporu v rámci projektu VEGA č. 2/6117/26. NMR spektrá boli merané za
podpory Slovenského národného programu pre výskum a vývoj č.
2003SP200280203.
1. Kronek J., Lustoň J., Böhme F., Chem. Listy, 92,
475 (1998)
2. Culbertson B. M. Prog Polym Sci 27, 579
(2002)
3. Aoi K., Okada M., Prog Polym Sci, 21,
151 (1996)
4. Lustoň J.; Kronek J.; Böhme F., J. Polym Sci Part A: Polym
Chem, 44, 343 (2006)
5. Lustoň J., Kronek J., Böhme F., Komber H., Macromol. Symp.,
164, 105 (2001)
6. Kronek J., Lustoň J., Böhme F., Komber H., Macromol. Symp.,
170, 301 (2001)
POLYMERIZAČNÉ REAKCIE CYKLICKÝCH
IMINOÉTEROV
J. LUSTOŇ, J. KRONEK
Ústav polymérov SAV, Dúbravská cesta 9, 842 36 Bratislava,
Slovensko
Cyklické iminoétery patria medzi heterocyklické zlúčeniny,
ktoré
majú dva heteroatómy v cykle. Z nich absolutne prevažujú 2-oxazolíny,
ktoré obsahujú dusík a kyslík v polohách 1 a 3 a dvojitú v polohe 2 v
5-člennom kruhu. Tento kruh možno otvoriť reakciou s nukleofilmi a tak
reaciou s aromatickými amínmi vzniká amido imínová štruktúra, s fenolmi
amido éterová väzba, s karboxylovými skupinami vzniká ester amid a s
tiolmi sa tvorí amido tioéterová jednotka. Okrem toho, 2-oxazolínová
skupina podlieha katiónovej polymerizácii, pričom je odolná voči
radikálovému ataku. To znamená, že vinylové alebo iné nenasýtené
deriváty 2-oxazolínov možno radikálovo polymerizovať so zachovaním
heterocyklickej štruktúry a katiónovo polymerizovať so zachovaním
nenasýtených väzieb. V nasledujúcom kroku možno zvyšné funkčné skupiny
použiť na modifikáciu, alebo sieťovanie. Použitím vhodného terminačného
činidla pri katiónovej polymerizácii alebo kopolymerizácii sa získajú
makroméry s rozličnými funkčnými skupinami. Na druhej strane, použitím
iniciátora s funkčnou skupinou možno získať telechelické polyméry,
ktoré sa môžu využiť na prípravu sekvenčných kopolymérov alebo
blokových kopolymérov. Zatial posledným druhom v tomto slede je
príprava hypervetvených polymérov a dendrimérov na báze 2-oxazolínov,
ktoré sa pripravujú z monomérov typu ABx, ktoré
je samozrejme nutné cielene pripraviť.
Autori ďakujú Slovenskej grantovej agentúre
za finančnú podporu v rámci projektu VEGA č. 2/6117/26
POLYMERNÍ KANCEROSTATIKA PRO PASIVNÍ
SMĚROVÁNÍ DO PEVNÝCH NÁDORŮ
T. ETRYCHa, P. CHYTILa,
T. MRKVANb, M. ŠÍROVÁb,
B. ŘÍHOVÁb, K. ULBRICHa
a Ústav makromolekulární chemie,
Akademie věd České republiky, Heyrovského n. 2, 162 06, Praha 6, Česká
republika,
b Mikrobiologický ústav, Akademie věd
České republiky, Vídeňská 1083, 142 20, Praha 4, Česká republika
email: etrych(imc.cas.cz
ÚVOD
V nedávné době bylo v naší laboratoři prokázáno, že syntetické
vodorozpustné kopolymery na bázi N-(2-hydroxypropyl)methakrylamidu
(HPMA) obsahující protinádorové léčivo doxorubicin (DOX) navázané přes
hydrolyticky degradovatelnou spojku přestavují potenciální systém pro
cílenou dopravu léčiv do modelových nádorů u myší1.
Ve
zmíněných konjugátech je léčivo napojeno na polymerní nosič na bázi
HPMA přes spojku obsahující hydrolyticky degradovatelnou hydrazonovou
vazbu. Tento polymerní systém je stálý ve vodném roztoku modelujícím
fyziologické prostředí (pH 7,4) a léčivo je uvolňováno při pH blízkém
prostředí endozomů a lyzozomů cílových buněk (pH 5 - 6). Předpokládáme,
že systémy neuvolňují během transportu v krevním řečišti aktivní
cytostatikum a že léčivo bude uvolněno ze systému po pinocytickém
vstupu do cílových buněk díky poklesu pH v endozomech (pH ~ 5-5.5)2.
HPMA kopolymery použité pro přípravu první generace výše uvedených
polymerních konjugátů byly vodorozpustné lineární polymery nebo
síťované polymery se zvýšenou molekulovou hmotností. Zvýšená
molekulární hmotnost síťovaných polymerních konjugátů vedla ke zvýšení
terapeutického účinku těchto systémů, který může být připsán tzv. "EPR
efektu" (enhanced permeability and retention effect) - zvýšené
akumulaci polymerního konjugátu s vyšší molekulovou hmotností (nad
limitem renální filtrace) v solidních nádorech. V tomto sdělení
prezentujeme syntézu a vlastnosti nových výše-molekulárních roubovaných
konjugátů na bázi kopolymerů HPMA připravených s cílem zvýšit jejich
záchyt v pevných nádorech pomocí EPR efektu. V těchto polymerních
konjugátech je hlavní řetězec nosného náhodného kopolymeru roubován
různými typy semitelechelických polymerů s cílem připravit polymerní
nosiče s vyšší molekulovou hmotností. Z důvodu zajištění eliminace
polymerního konjugátu jsou semitelechelické polymery připojeny k nosným
polymerům přes biodegradovatelné spojky podléhající enzymatické nebo
reduktivní intracelulární degradaci. Po intracelulární degradaci spojek
jsou degradační produkty polymerních konjugátů eliminovány z těla
glomerulární filtrací.
VÝSLEDKY A DISKUZE
Syntéza roubovaných polymerních konjugátů se skládá ze tří,
respektive čtyř syntetických kroků. V prvním kroku byly připraveny
kopolymery HPMA s methakrylovaným thiazolidin-2-thionem (TT)
oligopeptidu GlyPheLeuGly nebo methakrylovaným hydrazidem
6-aminohexanové kyseliny. V druhém kroku byly TT skupiny nosného
polymeru nechány reagovat s ethylendiaminem a vzniklé primární
aminoskupiny byly následně v některých případech modifikovány
2-iminothiolanem s cílem zavézt na nosný polymer thiolové skupiny. Ve
třetím kroku byly nosné polymery obsahující primární amino, hydrazidové
nebo thiolové skupiny roubovány semitelechelickými HPMA kopolymery
obsahujícími koncové reaktivní skupiny - sukcinimidylové estery,
thiazolidine-2-thionové skupiny nebo dithiopyridylové skupiny. V
posledním kroku byl na roubované polymerní nosiče obsahující v bočních
řetězcích hydrazidové skupiny navázán Dox.
Výsledky měření hydrolytického uvolňování léčiva in
vitro
ukázaly, že všechny roubované polymerní konjugáty s léčivem jsou
poměrně stabilní při 37 °C v pufru o pH 7,4. Po 24 hodinách inkubace
bylo zjištěno méně než 8 % uvolněného Dox, zatímco v pufru o pH 5 bylo
uvolněno za 24 h okolo 90 % Dox (37 °C). Nárůst molekulové hmotnosti
roubovaných polymerních konjugátů neměl významný vliv na rychlost
uvolňování léčiva, která zůstala obdobná jako u lineárních polymerních
konjugátů s nižší molekulovou hmotností.
Experimenty modelující degradaci roubovaných polymerních
konjugátů
byly prováděny ve fosfátovém pufru (pH 6) v přítomnosti kathepsinu B
nebo glutathionu jako degradačních činidel. Konjugáty obsahující
enzymaticky štěpitelné GlyPheLeuGly sekvence byly během 24 h
degradovány v přítomnosti kathepsinu B na degradační produkty s
molekulovou hmotností pod limitem renální filtrace. V roztoku
obsahujícím činidlo glutathion (3.10-6 mol/l)
jsme
pozorovali rychlý rozpad konjugátů obsahujících spojku náchylnou k
reduktivní degradaci. Také v tomto případě byly roubované polymerní
konjugáty rozštěpeny během 24 h na degradační polymerní produkty s
molekulovou hmotností okolo 25 000 g/mol.
Testy in vivo protinádorové aktivity byly
provedeny na myších
nesoucích T-buněčný lymfom EL4 v experimentálním uspořádání simulující
terapeutický režim. Výsledky experimentů prokázaly významný nárůst
protinádorového efektu roubovaných konjugátů oproti lineárním
polymerním konjugátům a také oproti účinku volného léčiva. Při vhodném
dávkování roubovaných polymerních konjugátů bylo dosaženo dokonce
vyléčení pokusných zvířat, a to až do 100% úspěšnosti. Uvedené výsledky
ukazují na široké možnosti, které polymerní léčiva nabízejí v boji s
jinak jen velmi obtížně léčitelnými chorobami.
Poděkování: Tato práce byla podporována
fa.
ZENTIVA a.s., MPO ČR (grant č. FI-IM2/111) a GA AV ČR (grant č.
A4050201).
Literatura:
- Etrych, T; Jelínková, M; Říhová, B; Ulbrich, K. J.
Controlled Release 2001, 73, 89.
- Etrych, T; Chytil, P; Jelínková, M; Říhová, B; Ulbrich, K. Macromol.
Biosci. 2002, 2, 43
- Ulbrich, K; Etrych, T; Chytil, P; Jelínková, M; Říhová, B. J.
Controlled Release 2003, 87, 33.
Polymerní nosiče léčiv s micelární
strukturou
P. Chytil1, T. Etrych1,
M. Šírová2, T. Mrkvan2,
B. Říhová2, K. Ulbrich1
1Ústav makromolekulární chemie Akademie
věd České republiky, Heyrovského nám. 2, 162 06, Praha 6
2Mikrobilogický ústav Akademie věd
České republiky, Vídeňská 1083, 142 20, Praha 4
e-mail: chytil(imc.cas.cz
Úvod
Nízkomolekulární cytostatika běžně používaná pro léčbu
nádorových
onemocnění mají řadu nevýhodných vlastností. Často jsou špatně
rozpustná ve vodě, díky malé specifitě účinku vykazují nežádoucí
vedlejší účinky, rychle se vylučují z organizmu a jejich biologická
využitelnost je malá. S cílem výrazně zvýšit účinek léčiv a odstranit
jejich nežádoucí projevy byla v posledních 30 letech navržena a
připravena řada různých polymerních nosičů léčiv. V naší skupině byly
syntetizovány konjugáty léčiva doxorubicinu (DOX) s vodorozpustnými
kopolymery na bázi N-(2-hydroxypropyl)methakrylamidu
(HPMA).
Pro vazbu léčiva na polymerní nosič byla využita kovalentní
pH-senzitivní hydrazonová vazba (1,2). Zatímco je tato vazba relativně
stabilní v pufru o neutrálním pH (model pH podmínek při transportu
konjugátu krevním řečištěm), v mírně kyselém pufru (pH 5 - 6) dochází k
uvolňování léčiva (model pH prostředí uvnitř endosomů buněk). Již dříve
jsme ukázali, že se tyto konjugáty vyznačují výrazným terapeutickým
efektem při léčbě EL4 lymfomu u myší (jedná se o agresivní nádorovou
linii leukemického typu). Zatímco léčba volným doxorubicinem je jen
málo účinná, v případě "hydrazonových" konjugátů při vhodném dávkování
lze dosáhnout 100% vyléčení pokusných zvířat (3).
Dále bylo prokázáno, že se vysokomolekulární látky, mezi nimi
i
syntetické kopolymery akumulují ve tkáni pevných nádorů v důsledku EPR
(enhanced permeability and retention) efektu více, nežli molekuly s
nižší molekulovou hmotností (4). Na základě tohoto poznatku jsme
navrhli vysokomolekulární micelární systémy, připravené na bázi
kopolymerů HPMA s DOX vázaným na nosič hydrolyticky štěpitelnou
hydrazonovou vazbou. V této studii prezentujeme syntézu a
fyzikálně-chemickou charakterizaci "hydrazonových" konjugátů, které
obsahují nejen hydrofilní kopolymer, ale i různé hydrofobní
substituenty (5). Tyto konjugáty tvoří ve vodném prostředí
vysokomolekulární polymerní micely. Navíc mohou interagovat s
biologickými membránami nádorových buněk (právě díky interakcím
hydrofobních substituentů s lipofilními částmi membrán) a zefektivnit
tak transport konjugátu do cílových buněk.
Výsledky a diskuze
Pro přípravu HPMA kopolymerů nesoucích hydrofobní
substituenty
jsme
použili různé struktury a obsahy substituentů, a to podle jejich
hydrofobicity (dodecyl, oleoyl, cholesteryl), tvaru (lineární,
planární) a také podle typu vazby mezi hydrofobním substituentem a
polymerním nosičem (viz Obrázek 1). Hydrodynamické poloměry micel ve
fyziologickém roztoku nabývaly hodnot od 6 do 18 nm a distribuce
velikosti částic byla relativně úzká. Také zdánlivé molekulové
hmotnosti těchto micel se pohybovaly v rozmezí 0,5 - 3,5 · 105,
přičemž molekulová hmotnost jednoho polymerního řetězce činila
přibližně 2 · 104.
Čím vyšší byly obsah a hydrofobicita substituentu, tím vyšší pak byly
zdánlivá molekulová hmotnost a hydrodynamický poloměr micel.
Uvolňování DOX z micelárních nosičů léčiv bylo ověřeno ve
fosfátových pufrech o pH 5 a 7,4 při 37 oC.
Všechny micelární systémy uvolňovaly DOX s rychlostí přibližně 50 %
DOX/24 h při pH 5 a 5 % DOX/24 h při pH 7,4. To znamená, že micelární
nosiče byly relativně stabilní - uvolňovaly jen minimum volného léčiva
- v pufru modelujícím pH krve. Na druhou stranu, v pufru modelujícím
vnitrobuněčné prostředí, docházelo k poměrně rychlému uvolňování DOX.
Porovnali jsme rychlost uvolňování DOX z micelárních nosičů s rychlostí
uvolňování z lineárního polymeru neobsahujícího hydrofobní
substituenty. Snížení rychlosti uvolňování z micelárních nosičů o 20 %
bylo zřejmě způsobeno stérickými zábranami v kompaktnější micelární
částici. Přesto tato rychlost zůstává dostatečná k tomu, aby množství
cytostatika uvolněného v buňce zajistilo výrazný cytotoxický efekt.
Biologický účinek micelárních konjugátů doxorubicinu byl
sledován jak v in vitro, tak v in vivo
podmínkách. Cytotoxicita konjugátu testovaná na řadě buněčných
nádorových linií (EL4 myší lymfom, BCL1 myší leukémie, myší lymfom
38C13, linie RAJI, 3T3 apod.) byla vysoká, srovnatelná s cytotoxicitou
lineárních polymerních konjugátů doxorubicinu. Obecně jsou kopolymery
tvořící ve vodném prostředí micely amfifilní povahy, často proto
projevují cytotoxické účinky, neboť mohou narušovat strukturu membrán
buněk. Nicméně námi připravené kopolymery bez léčiva nevykazovaly žádné
známky toxických účinků.
Pro posouzení vhodnosti systému jakožto léčiva je
nejdůležitějším testem in vivo
protinádorová aktivita. Ukázali jsme, že micely obsahující cholesteryl
jsou vysoce účinné proti myšímu EL4 lymfomu i při velmi malém dávkování
(1x10 nebo 2x5 mg ekvivalentu DOX/kg váhy myši, konjugát byl injikován
až po zřetelném vytvořené nádoru). Léčba tímto micelárním léčivem vedla
k úplnému vyléčení 100 %, resp. 88 % myší. Aplikace micel obsahujících
oleoyl (při stejném dávkování) nebo konjugátu bez hydrofobních
substituentů (1x15 mg DOX/kg) vedla k prodloužení doby života a u 20 %,
resp. 50% myší k úplnému vyléčení, protinádorové účinnosti micel s
cholesterolem však nedosáhla.

Obrázek 1 Schéma struktury micelárních
nosičů léčiv
Poděkování:
Tato práce byla podporována fa. Zentiva a.s., Ministerstvem
průmyslu
a obchodu (grant č. FI-IM2/111) a Grantovou agenturou Akademie věd
České Republiky (grant č. IAA4050201).
Reference:
1. Etrych T. et al., Macromol. Biosci. 2,
43-52 (2002)
2. Ulbrich K. et al., J. Controlled. Release
87, 33-47 (2003)
3. Mrkvan T. et al., J. Controlled Release
110, 119-129 (2005)
4. Maeda H. et al., Crit. Rev.
Ther. Drug Carrier Syst. 6, 193 (1989)
5. Patent No. CZ PV 2006-207
SULFENAMID - NOVÝ TYP REDUKTIVNĚ ŠTĚPITELNÉ
VAZBY PRO DOPRAVU A ŘÍZENÉ UVOLŇOVÁNÍ LÉČIV
M. STUDENOVSKÝ, K. ULBRICH
Ústav makromolekulární chemie AV ČR, Heyrovského nám. 2, 162
06 Praha 6, Česká Republika
studenovsky(imc.cas.cz
SOUHRN:
Sulfenamidová vazba byla navržena jako snadno thiolyticky
štěpitelné
propojení polymerního nosiče s aktivním léčivem. Bylo navrženo a
připraveno několik modelových sulfenamidů s různou chemickou strukturou
s cílem dosáhnout snadné štěpitelnosti intracelulárním glutathionem za
dostatečné stability v krevním řečišti.
ÚVOD:
Zatímco většina léčiv v čisté podobě vykazuje řadu negativních
vlastností (toxicita, nízká specificita, krátká doba cirkulace v krvi),
jejich zabudování do polymerních konjugátů může tyto nedostatky
odstranit. Z těchto důvodů je v posledních desetiletích vývoji
polymerních léčiv věnováno značné úsilí.
Polymerní léčivo lze posuzovat z mnoha hledisek, zásadní roli
však
hraje samotné nízkomolekulární léčivo, polymerní nosič a způsob
vzájemného spojení. V současné době je k dispozici velké množství léčiv
vhodných pro přípravu polymerních konjugátů a rovněž řada osvědčených
biokompatibilních polymerů. Způsobů vzájemného propojení, splňujících
požadavky na stabilitu během transportu krví a snadnost štěpení v místě
cílení, však zatím bylo vyvinuto pouze několik. Principielně mohou být
rozděleny do tří skupin: 1. hydrolyticky labilní vazby
(pH senzitivní hydrazonová vazba); enzymaticky degradovatelné vazby
(Gly-Phe-Leu-Gly peptidová sekvence); 3. reduktivně štěpitelné vazby
(disulfidová vazba).
Zatímco pH senzitivní spojení obvykle vykazuje nedostatečnou
selektivitu a enzymaticky degradovatelné vazby na druhé straně značnou
citlivost na aktivitu intracelulárních enzymů, využití redukčního
prostředí uvnitř buňky se zdá být elegantním řešením. Dosud jediným
používaným systémem tohoto druhu je disulfidová vazba, která je však
omezena na úzkou skupinu léčiv obsahujících SH skupinu. Využití častěji
se vyskytujících funkčních skupin v molekule léčiva (např. aminoskupin)
by značně rozšířilo paletu potenciálních léčiv. Sulfenamidová
vazba (-S-NH-) tuto možnost nabízí.
Cílem této práce je navržení a optimalizace syntézy
polymerních
konjugátů na bázi reduktivně štěpitelné sulfenamidové vazby.
Potenciálně využitelná léčiva pak mohou být kterákoli ze skupiny
nesoucích primární či sekundární aminoskupinu (např. chemoterapeutika
Doxorubicin, Daunorubicin,
Raltitrexed). Obrázek 1 ukazuje předpokládaný mechanismus
vázání
léčiva na polymerní nosič a jeho uvolňování v prostředí
intracelulárního glutathionu.

Obr. 1 (a) syntéza polymerního konjugátu; (b) uvolnění volného
léčiva účinkem glutathionu. X = Cl, Br příp. ftalimido skupina.
První fáze projektu zahrnovala návrh, syntézu a
charakterizaci
dvou
modelových nízkomolekulárních sulfenamidů, v nichž je léčivo
reprezentováno molekulou benzylaminu (Obr. 2)
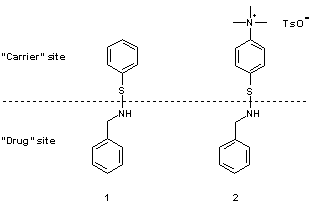
Obr. 2 Struktura modelových sulfenamidů a jejich funkce v
"drug delivery" systému. TsO- = 4-toluensulfonát.
POUŽITÉ METODY:
Všechny použité chemikálie byly zakoupeny od společností Fluka
a
Aldrich v dostatečné čistotě. Jednotlivé syntézy zahrnovaly standardní
procedury organické chemie. Charakterizace připravených látek byla
provedena pomocí elementární analýzy, NMR a HPLC.
Sulfenamidy 1 a 2 byly
připraveny vícekrokovými
syntézami. Jejich hydrolytická stabilita a kinetika thiolýzy byla
měřena při pH 7.5 a 5.1 při koncentraci glutathionu 5 mM. Měření byla
prováděna při 37 °C se vstupní koncentrací sulfenamidu 3 mM.
Kvantitativní údaje byly získány pomocí HPLC chromatografie.
VÝSLEDKY A DISKUSE:
Rychlost thiolýzy a hydrolytická stabilita byly určeny jako
časová
závislost konverze výchozích sulfenamidů a uvolněného benzylaminu.
Obrázek 3 ukazuje velmi rychlé štěpení 1
glutathionem. Hydrolytická stabilita tohoto modelového sulfenamidu však
byla poměrně nízká (Obr. 4).

Obr. 3 Štěpení 1 účinkem glutathionu při
pH 5.1 (intracelulární prostředí).
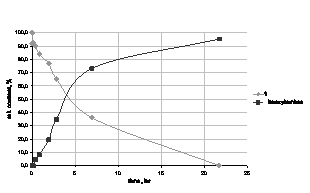
Obr. 4 Hydrolytický rozklad 1 při pH 7.5
(krevní řečiště).
Vzhledem k tomu, že náchylnost sulfenamidové vazby k hydrolýze
je dána bazicitou dusíkového atomu, byl připraven derivát 2,
v němž je bazicita atomu dusíku dramaticky snížena přítomností
trimethylammoniové skupiny. Kinetická měření však oproti očekávání
neukázala významné zvýšení stability. Derivát 2
vykazoval obdobnou rychlost štěpení glutathionem i náchylnost k
hydrolýze, jako sulfenamid 1.
Možností zvýšení hydrolytické stability sulfenamidu další
změnou
jeho chemické struktury existuje mnoho. Byly navrženy nové typy
modelových struktur, jež jsou v současné době studovány.
ZÁVĚRY:
Sulfenamid je nadějným typem labilní vazby mezi léčivem a jeho
nosičem. Umožňuje extrémně rychlé štěpení v redukčním prostředí uvnitř
buňky a při vhodné chemické modifikaci i dostačující stabilitu v
krevním řečišti. Tyto vlastnosti předurčují sulfenamidovou vazbu pro
další studium a využití pro design polymerních léčiv.
LITERATURA:
Pillai
O., Panchagnula R.: Polymers in drug delivery, Curr. Opin. Chem.
Biol., 5 (2001) 447-451.
Hoste
K., De Winne K., Schacht E.: Polymeric prodrugs, Int. J. Pharm.,
277 (2004) 119-131.
Ulbrich
K., Šubr V.: Polymeric anticancer drugs with pH-controlled
activation, Adv. Drug Delivery Rev., 56 (2004) 1023-1050.
Fontana
A., Marchiori
F., Moroder L., Scoffone E.: New removal conditions of sulfenyl grous
in peptide synthesis, Tetrahedron Lett., 26 (1966) 2985-2987.
PODĚKOVÁNÍ:
Tento projekt je financován Grantovou agenturou České
Republiky, grant č. 204/05/2255.
MULTIBLOKOVÉ POLYMERY NA BÁZI PEG,
OBSAHUJÍCÍ HYDROLYTICKY A REDUKTIVNĚ ŠTĚPITELNÉ VAZBY
A. BRAUNOVÁ, M. PECHAR, K. ULBRICH
Oddělení biomedicinálních polymerů, Ústav makromolekulární
chemie
AVČR, Heyrovského nám. 2, 162 06 Praha 6, Česká republika
(braunova(imc.cas.cz)
Poly(oxyethylen)y (PEG) jsou velmi dobře definovatelné,
jednoduché a
snadno dostupné vodorozpustné polymery s vlastnostmi, vhodnými pro
různorodé aplikace v chemii, biotechnologii i v medicíně. Navíc se PEG
vyznačuje velmi dobrou rozpustností jak v polárních, tak i nepolárních
rozpouštědlech. Kovalentním navázáním PEG na biologicky aktivní látky
(BAL, např. hydrofobní léčiva, proteiny, vektory pro genovou terapii)
lze tedy docílit změny povrchových vlastností BAL a jejich lepší
rozpustnosti ve vodném prostředí, omezení povrchové adsorpce proteinů,
usnadnění transportu molekul přes buněčné membrány a zdokonalení jejich
farmakokinetiky. Modifikace poly(oxyethylen)em je vzhledem k uvedeným
vlastnostem hojně využívána jako metoda pro redukci různých nežádoucích
následků biologického rozpoznání BAL, reprezentovaných imunogenicitou a
antigenicitou v případě proteinů, rychlým zachycením liposomů orgány
retikuloendotheliálního systému a trombogenicitou, buněčnou přilnavostí
a proteinovou adsorpcí v případě umělých biomateriálů.
V této práci byly navrženy, připraveny a studovány vlastnosti
nových
vysokomolekulárních multiblokových multivalentních polymerů PEG s
deriváty L-cysteinu, resp. cystinu či cystaminu, obsahující v hlavním
řetězci hydrolyticky degradovatelné esterové nebo urethanové vazby a
zároveň redukci (vlivem redukčního činidla glutathionu) podléhající
disulfidové můstky. Ve většině případů byla nejprve provedena modelová
studie biodegradace polymeru na diblokových polymerech, obsahujících
bloky semitelechelického monomethoxy-poly(oxyethylen)u (mPEG) a
příslušné spojky. Degradační studie byly prováděny v prostředí
modelujícím podmínky v živém organismu (pH 5,0 - 5,5 ~ lysosomy, pH 7,4
~krevní řečiště, pH 7,4 - 8,0 ~ trávicí trakt, glutathion = GSH
~cytoplasma). Struktura blokového polymeru byla navržena tak, aby
spojka obsahovala funkční skupiny umožňující buď vazbu biologicky
aktivní molekuly (cytostatika, proteiny, enzymy), nebo připojení
multiblokového polymeru k povrchu nanočástic typu polykationtového DNA
komplexu nebo viru, za současného zachování biodegradability této
spojky.
Byl vypracován postup oxidační polykondenzace (viz obrázek 1),
kterým lze reprodukovatelně připravit tyto multiblokové polymery s
vysokou molární hmotností a poměrně úzkou distribucí molekulových
hmotností. Polymer 1 byl následně modifikován buď sukcinanhydridem nebo
N-sukcinimidyl-3(2-pyridyldithio)propionátem (SPDP)
a
3-sulfanylpropionovou kyselinou a v obou případech aktivován až na
reaktivní nitrofenylové estery - polymer 2, resp.3 (viz obrázek 2),
které mohou být použity např. pro povrchovou modifikaci komplexů
poly-L-lysin/DNA.
Připravené blokové polymery byly studovány jak z hlediska
vlivu
různých inkubačních prostředí, modelujících přirozené prostředí v
organismu (odlišné pH, přítomnost GSH) na rychlost štěpení
degradovatelných vazeb, zavedených do struktury polymerů, tak i z
hlediska vlivu jednotlivých koncových skupin v bočních řetězcích
polymerů na rychlost degradace jednotlivých polymerních derivátů. Bylo
zjištěno, že se zvyšujícím se pH (v rozmezí pH 5,5 - 8,0) se stabilita
degradovatelných esterových vazeb snižuje. Redukce zavedených
disulfidových vazeb v přítomnosti fyziologické koncentrace GSH (1 - 5
mM) probíhá u všech připravených derivátů významnou rychlostí. Zároveň
lze konstatovat, že změnou struktury koncových skupin bočních řetězců
či zavedením dalších reduktivně štěpitelných S-S můstků do bočního
řetězce lze řídit rychlost degradace celého polymerního systému. Po
případné konjugaci těchto multiblokových polymerů s BAL lze očekávat
prodloužení doby cirkulace konjugátů polymer-BAL v organismu a
následnou poměrně specifickou degradaci v cytoplasmě cílových buněk,
což je velmi výhodné zvláště u polymerem modifikovaných vektorů pro
dopravu DNA.
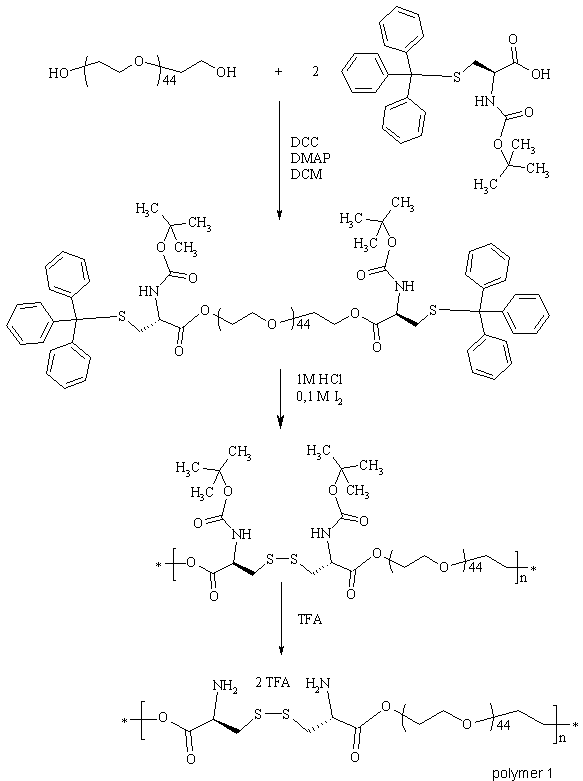
Obrázek 1 Syntéza multiblokového polymeru na
bázi PEG-cystinu s degradovatelnou esterovou a disulfidovou vazbou
Připravené vysokomolekulární biodegradovatelné multiblokové
polymery tedy výhledově představují nový typ polymerních nosičů
cytostatických léčiv. V případě biologicky aktivních proteinů či DNA by
modifikace polymerem měla zabránit předčasnému odstranění těchto
systémů z krevní plasmy a z organismu a ochránění jejich aktivních míst
před biologickou degradací. Navíc použití zmíněných multiblokových
multivalentních polymerů nabízí i možnost směrování jak polymerních
léčiv, tak i vektorů pro dopravu DNA.
|

|
|
| Textové pole: Obrázek 2 Polymer 1 modifikovaný na
aktivní nitrofenylové estery |
|
Autoři
děkují Ministerstvu školství mládeže a tělovýchovy za podporu projektu
grantem č.IM4635608802.
Biodegradovatelné termoresponsivní polymery
pro biomedicinální aplikace
M. Hrubýa, J. Kučkab,
H. Mackováa, M. Babiča,
K. Ulbricha, L. Lešetickýc,
O. Lebedab
aÚstav makromolekulární chemie,
Akademie věd České republiky, Heyrovského nám. 2, 162 06 Praha 6, Česká
republika
bÚstav jaderné fyziky, Akademie věd
České republiky, 250 68 Řež u Prahy, Česká
republika
cPřírodovědecká fakulta, Univerzita
Karlova, Hlavova 2030, 128 43 Praha 2, Česká repulika
*adresa pro korespondenci: mhruby(centrum.cz
(M. Hrubý)
Termoresponsivní (někdy take nazývané termosensitivní)
polymery, jako jsou například poly(N-isopropylakrylamid),
poly(N-isopropylmethakrylamid),
peptidové sekvence odvozené od struktury bílkoviny elastinu
("elastin-like" peptidy) nebo poly(ethylenoxid-co-propylenoxid),
jsou rozpustné ve vodě při laboratorní teplotě a při překročení dolní
kritické rozpouštěcí teploty (LCST) dojde k jejich fázové separaci z
roztoku. Pokud je jejich LCST blízko teploty lidského těla, mohou
představovat slibné materiály pro biomedicinální použití [1], například
jako injikovatelná lokální depa léčiv, nosiče léčiv cílené lokální
hypertermií, termoresponsivní micely pro cílený transport a řízené
uvolňování léčiv, injikovatelné implantáty atd.
Fenomén teplotně závislé fázové separace polymeru z vodného
roztoku
je způsoben soutěžením teplotně závislých sil rozpouštějících polymer
(solvatace polymerního řetězce a tvorba vodíkových vazeb mezi polymerem
a vodou) a hydrofóbními interakcemi, které vedou k fázové separaci
polymeru z roztoku. Pokud je tedy do kopolymeru zabudován komonomer,
který je polárnější, LCST se posouvá k vyšším teplotám a naopak. Je-li
do kopolymeru zabudován komonomer, který je hydrofóbnější než základní
polymer, ale podléhá hydrolytické degradaci a tím se přemění na
hydrofilní strukturu, pak LCST takového kopolymeru stoupá v průběhu
hydrolýzy.
Lokální aplikace termoresponsivního polymeru s LCST mezi
laboratorní
a tělesnou teplotou nesoucího radionuklid jako systému pro lokální
radioterapii má některé výhody. Polymer může být označen a aplikován do
místa určení jako kapalný vodný roztok injekčně bez nutnosti
chirurgické implantace, a na místě aplikace dojde k jeho fázové
separaci teplotou lidského těla a tím i tvorbě lokálního depa.
Radionuklid by tak neměl být vyplavován dále do těla. Pokud je LCST
takového polymeru nastavena mezi laboratorní teplotu a teplotu lidského
těla pomocí kopolymerace hydrofóbního komonomeru, který při postupné
hydrolytické degradaci v lidském těle přechází na hydrofilní strukturu
a posouvá tak LCST nad teplotu lidského těla, polymer se biodegradací
rozpouští, což by mělo urychlit odstranění takového polymeru po rozpadu
radionuklidu z organismu. Aplikace takového systému by byla možná
například pro radiosynovektomie v kloubních aplikacích (kde se již
využívají lokální radioterapeutika, např. koloidní 198Au),
nebo jako pomocná radioterapie po chirurgické resekci nádorové tkáně.
Takový degradovatelný termoresponsivní polymerní systém by mohl najít
uplatnění i pro další výše zmiňované biomedicinální aplikace.
Byly připraveny a charakterizovány kopolymery N-isopropylmethakrylamidu
(LCST homopolymeru 44 °C, Mw
kopolymerů 30 - 40 kDa) s 3 různými komonomery obsahujícími hydrofóbní
skupiny 3 různých velikostí a tím i hydrofobicit (C3,
C6 a C12) vázané
hydrolyticky labilní hydrazonovou vazbou. Do polymeru byly rovněž
vneseny skupiny značitelné 131I (tyrosinamid)
respektive 90Y
(1,4,7,10-tetraazacyclododekan-1,4,7,10-tetraoctová kyselina; DOTA).
Chování kopolymerů ve vodném roztoku je silně závislé na použitém
degradovatelném hydrofóbním komonomeru, při použití monomeru s C12
zbytkem polymer tvoří ve vodném prostředí při teplotě pod LCST micely,
které pak postupně kolabují až do makroskopické fázové separace. Při
použití monomeru s C6 zbytkem je závislost LCST
na obsahu
hydrofóbního monomeru lineární a LCST těchto kopolymerů může být
nastaveno poměrem monomerů v rozmezí 13 - 37 °C před degradací a 44 -
47 °C po hydrolytické degradaci. Monomer s C3
zbytkem už je
příliš hydrofilní a LCST zvyšuje. Za modelových podmínek při 37 °C ve
vodném pufru o pH 7,4 bylo prokázáno kompletní rozpuštění separované
fáze během 7 dnů.
Studie byla vypracována za finanční podpory Grantové agentury
Akademie věd České republiky (grant č. A400480616) a programu
výzkumných center Ministerstva školství, mládeže a tělovýchovy České
republiky (grant č. IM 4635608802).
LITERATURA
[1] Chilkoti, A., Dreher, M. R., Meyer, D. E., Raucher, D.: Adv.
Drug Deliv. Rev. 2002, 54(5), 613-630.
Nové polymerní nosiče 198Au
A 64Cupro radioterapii
J. Kozempela,b, M. Hrubýc*,
J. Kučkaa,b, O. LebeDAa
aÚstav jaderné fyziky, Akademie věd
České republiky, 250 68 Řež u Prahy, Česká
republika
bUniverzita Karlova, Přírodovědecká
fakulta, Katedra organické a jaderné chemie, Hlavova 2030, 128 43 Praha
2, Česká republika
cÚstav makromolekulární chemie,
Akademie věd České republiky, Heyrovského nám. 2, 162 06 Praha 6, Česká
republika
*adresa pro korespondenci: mhruby(centrum.cz
(M. Hrubý)
Mezi isotopy prvků I. B skupiny periodické tabulky je několik b-
zářičů výhodných pro radioterapii. Patří mezi ně především 198Au
(poločas rozpadu T1/2 = 2,7
dne) a 64Cu (T1/2
= 12,7 hodiny, kromě b-
rozpadu se rozpadá i b+
rozpadem a elektronovým záchytem ).
Radioisotopy mají řadu výhod pokud jsou použity jako aktivní
složka
polymerních cílených terapeutik pro léčbu nádorových onemocnění.
Především hmotnost radioisotopu je často o několik řádů nižší (zejména
u tzv. beznosičových preparátů), než je efektivní dávka chemického
kancerostatika. Radioisotop se dále nemusí uvolnit z polymerního
nosiče, aby byl efektivním při zabíjení nádorových buněk. Na druhou
stranu je ovšem třeba zmínit i nevýhody radioterapeutik, kde je hlavním
problémem radiační zátěž zdravé tkáně během transportu na místo určení
a s tím související požadavek na rychlý transport systému do cílové
tkáně.
Pro přípravu polymerního systému pro celkovou či lokální
radioterapii je třeba radionuklid navázat na polymerní nosič vazbou,
která je dostatečně stabilní i v biologickém prostředí. Pro výše
uvedené ionty je pro tyto účely nejužívanějším ligandem DOTA ([1],
1,4,7,10-tetraazacyclododekan-1,4,7,10-tetraoctová kyselina), která je
velmi silným komplexačním činidlem, ale má některé nevýhody.
 ¨
¨
Obrázek 1. Struktury
chelatujících ligandů pro 198Au3+
a 64Cu2+.
Nevýhodou DOTA jako ligandu je především obtížná syntetická
dostupnost některých jejích vhodných derivátů pro definovanou
funkcionalizaci polymerů a dále směrující účinek tohoto ligandu do
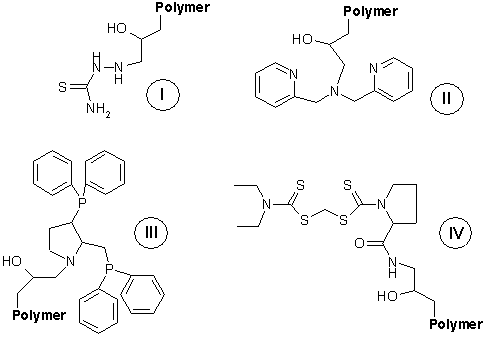
ledvin díky přítomnosti karboxylových skupin [1]. Proto jsme se v této
studii zaměřili na možnost využití jiných komplexujících skupin pro
vazbu 198Au3+
a 64Cu2+
na polymerní nosič (viz Obrázek 1). Jako modelový vodorozpustný
biokompatibilní polymer jsme zvolili poly{[N-(2-hydroxypropyl)methakrylamid]-co-glycidylmethakrylát}
(Mw
= 120 kDa, glycidylmethakrylát 7,5 molárních procent monomerních
jednotek), obsahující reaktivní epoxidové skupiny, za které byly
navázány vybrané chelatující skupiny (viz Obrázek 1).
Polymer I
byl připraven reakcí epoxidového prekurzoru s hydrazinem a následnou
reakcí vzniklého alkylhydrazinu s rhodanidem amonným, polymer II
reakcí epoxidového prekurzoru s dipikolylaminem, polymer III
aminolýzou epoxidového prekurzoru
3-(1,1-difenylfosfino)-2-[(1,1-difenylfosfino)methyl]pyrrolidinem a
polymer IV aminolýzou epoxidového prekurzoru
amoniakem a reakcí vznikého primárního aminu s činidlem IX
([(diethylamino)karbothioyl]sulfanylmethyl
2-[(2-thioxo-1,3-thiazolan-3-yl)karbonyl]-1-pyrrolidinkarbodithioátem).
Činidlo IX bylo připraveno podle schématu na Obrázku
2.
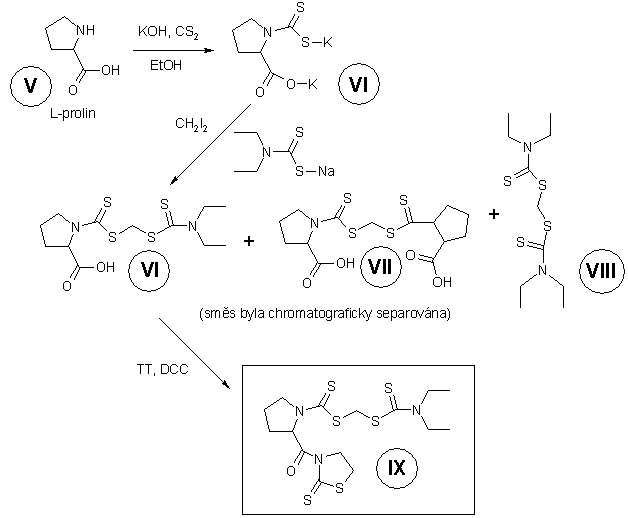
Obrázek 2. Příprava
činidla IX.
198Au a 64Cu
byly
připraveny v jaderném reaktoru Ústavu jaderného výzkumu, a.s., v Řeži u
Prahy ozařováním zlata respektive mědi v přírodním isotopickém
zastoupení neutrony 197Au (n,g)
198Au respektive 63Cu
(n,g) 64Cu
reakcí. Kovy byly poté převedeny na příslušné soli rozpuštěním kovů v
minimálním objemu směsi koncentrovaná kyselina dusičná - koncentrovaná
kyselina chlorovodíková (1:3 v/v) a převedením na chloridy konc. HCl.
Polymery I - IV byly označeny 198Au3+
a 64Cu2+
v pufrovaném prostředí a výtěžek isotopického značení byl stanoven po
oddělení makromolekulární frakce gelovou filtrací na odsolovací kolonce
PD-10. Nejvyšší výtěžek značení 198Au
i 64Cu byl dosažen s
polymerem I obsahujícím thiosemikarbazidové funkční
skupiny (>95 %), pro 198Au
byl prakticky kvantitativní výtěžek dosažen i při značení polymeru IV.
Dipikolylamin dobře komplexuje měďnaté ionty, ale výtěžek značení
zlatitými ionty je malý (cca 4 %). Ačkoliv je popisovaná značná
stabilita komplexů zlata s fosfinovými ligandy [2], výtěžek značení
polymeru III 198Au byl jen
15 %. Fosfinový ligand
zároveň způsobuje redukci zlatitých iontů na fialové koloidní zlato.
Pro další studie proto budou použity thiosemikarbazid jako ligand pro 198Au
a 64Cu a
methylenbis(dialkyldithiokarbamát) jako ligand pro 198Au.
Studie byla vypracována za finanční podpory Grantové agentury
Akademie věd České republiky, grant číslo A400480616.
LITERATURA
[1] Li, L., Yazaki, P. J., Anderson, A. L., Crow, D., Colcher,
D.,
Wu, A. M., Williams, L. E., Wong, J. Y. C., Raubitschek, A., Shively,
J. E.: Bioconjugate Chem. 2006, 17(1), 68-76.
[2] Berning, D. E., Katti, K. V., Volkert, W. A.,
Higginbotham, C. J., Ketring, A. R.: Nucl. Med. Biol.
1998, 25(6), 577-583.
BIOMATERIÁLY NA BÁZE PRÍRODNÝCH POLYMÉROV
D. BAKOŠa, S. BUBENÍKOVÁa,
L. VODNÁa, I. LACÍKb
aDepartment of Plastics and Rubber,
Faculty of Chemical
and Food Technology, Slovak Technical University, Radlinského 9,
Bratislava, Slovak Republic, dusan.bakos(stuba.sk
bPolymer Institute, Slovak Academy of
Science, Dúbravská
cesta 9, Bratislava, Slovak Republic, Centre of Excellence of SAS
“CEDEBIPO” for Degradation of Biopolymers
Introduction
Polyelectrolyte complexes based on natural polymers represent
an
attractive class of polymer-based materials finding an irreplaceable
role in preparation of biodegradable and biocompatible 3D membranes.
These membranes are effectively used as scaffolds in tissue engineering
for a replacement connective tissue. Numerous factors affect the
properties of the polyelectrolyte complex placing the stringent
requirements on the selected polyelectrolytes as well as on the
preparation conditions. From the point of view of polymer hydrogels,
the polyelectrolyte complexes belong to the category of the physically
crosslinked gels with the crosslinks of small but finite energy and/or
of finite lifetime. Usually, chemical crosslinking has been widely used
to increase the mechanical and biological stability of such
biomaterials. The clinical requirement for artificial graft materials
for promoting an effective wound repair is large (burn injuries, post
traumatic skin and soft tissue defects, pressure sores, diabetic skin
ulcers, venous stasis ulcers, and defects arising following tumor
excision). A matrix for biodegradable implants based on collagen,
hyaluronan or chitosan occupy prominent place in the research at
present. In our projects, we have studied several types of such
biomaterials – the collagen/hyaluronan and chitosan lactate/hyaluronan
membranes chemically modified with starch aldehyde derivatives. Here we
present newly elaborated chitosan-based microparticulate systems.
Chitosan-based microparticulate systems have been reported,
using
various encapsulation methods and chemistries, for a variety of
applications. Interactions between positively-charged amino groups and
negatively-charged counterions have been used to prepare microparticles
either by ionotropic gelation, or by polyelectrolyte complexation. The
properties of chitosan-based capsules depend on a number of parameters,
which role one needs to understand in order to control the capsule
formation process and capsule properties. The study is focused on
preparation of biodegradable chitosan microcapsules with tailored
properties for potential applications in medical field as protein and
cell temporary carriers. Using the multi-loop reactor to control
reaction time in the range of tens of seconds is absolutely crucial for
this capsule type.
Materials and methods
The following
polymers were used for capsule preparation: high viscosity chitosan,
CHIT, (SIGMA,viscosity ~ 400 mPa.s for 1 wt.% in 1 % aq. acetic acid at
20°C), sodium hexametaphospate, TPP, (SIGMA, MW 367.85) and chondroitin
sulphate 6, CHS6 (Lambda Life, cell culture tested). All other
chemicals were of analytical grade.
Capsule preparation and characterization
1 wt% CHIT solution was prepared by dissolving CHIT in 0.5 M
aqueous
acetic acid and pH was slowly increased to 5 with aqueous NaOH. CHIT
solution was air-stripped to multi-loop reactor [2], which was filled
by solution of 0.5 wt% CHS6 with 0.5 wt% TPP (pH 7). All solutions were
prepared in 0.9 wt% NaCl. Capsules were filtered, washed and stored in
0.9 wt% NaCl. The antibiotic ofloxacine was entrapped in chitosan
solution before complexation. The release of ofloxacin was studied
using UV/VIS spectrophotometric determina-tion at 293 nm.
The capsule diameter and membrane
thickness were
measured by optical microscopy (Kapa 2000, Kvant, Bratislava, SR) using
CCD camera (CC-63KW1P, Mintron Malaysia), by means of digital imaging
using Prover ImageForge 1.1 software.
Results and discussion
CHIT dropped into the polyanionic solution produced
immediately
capsules with a spherical shape, formed trough ionic interactions at
the interface of CHIT droplets. The internal diffusion coefficient De
was determined by the rate of diffusion of TPP into the chitosan gel,
using thickness of the membrane in expressing concentration changes in
a diffusion process. We experienced that concentration of TPP (lower
molecular polyanion) does not influence diffusion. On the other hand,
the presence of CHS6 changes the situation and this higher molecular
polyanion is more localised on the surface of the bead and has a strong
influence on mechanical properties. Other parameters, like pH and ionic
strength during the complex formation, concentration, gelation time as
well as ratio between TPP and CHS6 need to be optimized to form
non-sticky, spherical and chemically and mechanically stable core-shell
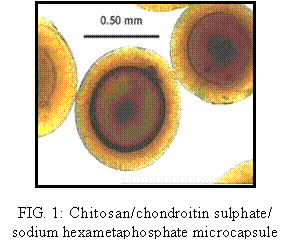
type microcapsules shown in Figure 1. In order to understand mutual
effects of several these factors on membrane thickness of beads, the
design of experiment method (DOE) was used.
|
|
 |
From the experiments it is clear that using the multi-loop
reactor
to control reaction time in the range of tens of seconds is absolutely
crucial for this capsule type. Having the standardized beads we are
able to control the release of antibiotics from the beads. The same
model as before we used for the calculation of the internal diffusion
coefficient De of effusion of ofloxacine from the standardized bead.
The 0.9% NaCl solution at the temperature 37 °C was used as solvent.
The amount of the antibiotic effused from the beads into the
surrounding solution was regularly measured and concentrations and De
were estimated. From the results, the release of ofloxacin was regular.
Acknowledgment
This work was supported by Science and
Technology Assistance Agency under the contract No. APVT-20-016002 and
20-015904.
BIODEGRADABLE POLYURETHANE FOAMS
L. VOJTOVÁa, J. JANČÁŘa, , L.BABÁKb, I. MÁROVÁb, J.DAVIDc, M. VÁVROVÁc
aInstitute of Materials Chemistry, Fakulty of Chemistry, Brno University of Technology, Purkyňova 118, CZ-612 00 Brno, Czech Republic (vojtova/fch.vutbr.cz, http://www.fch.vutbr.cz/)
bInstitute of Food Science and Biotechnology, Fakulty of Chemistry, Brno University of Technology, Purkyňova 118, CZ-612 00 Brno, Czech Republic
cInstitute of Chemistry and Technology of Environmental Protection, Fakulty of Chemistry, Brno University of Technology, Purkyňova 118, CZ-612 00 Brno, Czech Republic
Abstract
In order to avoid environmental
pollution by non-degradable polymer waste dump, new biodegradable
flexible polyurethane (BIO-PUR) foams modified by natural materials
with a controlled period of decomposition were investigated with
respect to their microbial degradation and ecotoxicological evaluation.
PUR foams are used widely in many fields
as structural, cushion, insulation, electrical, flotation and packing
materials. Commonly they are discarded after being used representing
serious contamination problems due to their difficult disintegration
and incorporation to the environment.
Flexible PUR foams are usually obtained
from the reaction between polyfunctional alcohol (polyol),
polyisocyanate and water as a blowing agent. The most widely used is
one-shot process, where direct mixing of coreactants and simultaneous
addition of water, catalyst and other additives are used. Foam
properties are affected by the properties of raw materials and also can
be modified by a wide variety of additives, such as fillers,
stabilizers, cross-linking agents and chain extenders1.
In addition, natural polymers containing more than one hydroxyl group
in the main chain (e.g. starch, cellulose, tannin, saccharide etc.) are
expected to be utilized as biodegradable polyols for PUR preparation2.
In this proposed work, flexible BIO-PUR
foams were prepared by one-shot process using tolylene diisocyanate
(TDI), polyether polyol (PEP), catalysts, water and 4 types of
cellulose or starch derivatives with different degree of substitution
(DS) of -OH groups such as: acetylated starch (DS = 0,1),
acetylcellulose (DS = 2,4), 2-hydroxyethylcellulose (DS = 0,6) and
carboxymethylcellulose sodium salt (DS = 0,7), AS, AC, CMC, HEC,
respectively. The added amount of bio-polyol substituted from 1 to 10
weight % of common PEP used in reference PUR (ref. PUR) foam. According
to the literature, using the cellulose or starch derivatives instead of
a natural cellulose or starch increases the reactivity of -OH groups
with diisocyanates and decreases the hardness of the prepared BIO-PUR
foams by opening the cells of PUR polymer network. Moreover,
replacement of conventional PEP up to 10 weight % should not affect the
mechanical properties of the original PUR foams. The phenomena
mentioned above are favorable mainly for the preparation of soft
flexible foams3.
The FT-IR characterization of BIO-foams
showed characteristic peaks for cellulose or starch derivatives and
polyurethane. There were not found any distinctive differences between
the BIO-PUR foams and the ref. PUR either by sight or by applying the
polarization microscope, where pore size and homogeneity of polymer
network were compared (FIG. 1), except for the BIO-PUR with AC. Only
the AC bio-polyol was not soluble in common PEP (due to the high DS)
and thus some small particles of powdered additive were observed in PUR
network via microscope.
| A |
 |
B |
 |
FIG. 1: Pictures of (A) Ref. PUR and (B) BIO-PUR, where 10 wt % of PEP was replaced by CMC.
Resulting BIO-PUR foams filled with AC, AS and HEC were undergone microbial degradation using the mixed culture of Thermophillus sp. and yeast strain Aureobasidium pullulans that
produce a large amount of hydrolytic enzymes capable of degrading
polymeric materials. 1 g of each sample of BIO-PUR foams was degraded
for 4 weeks. Each week 0.5 g of lactose was added into cultivation
medium and in regular intervals the concentration of biomass was
determined (g of cells /l of medium). After cultivation, surface
microscopy of polyurethanes was tested (FIG. 2).
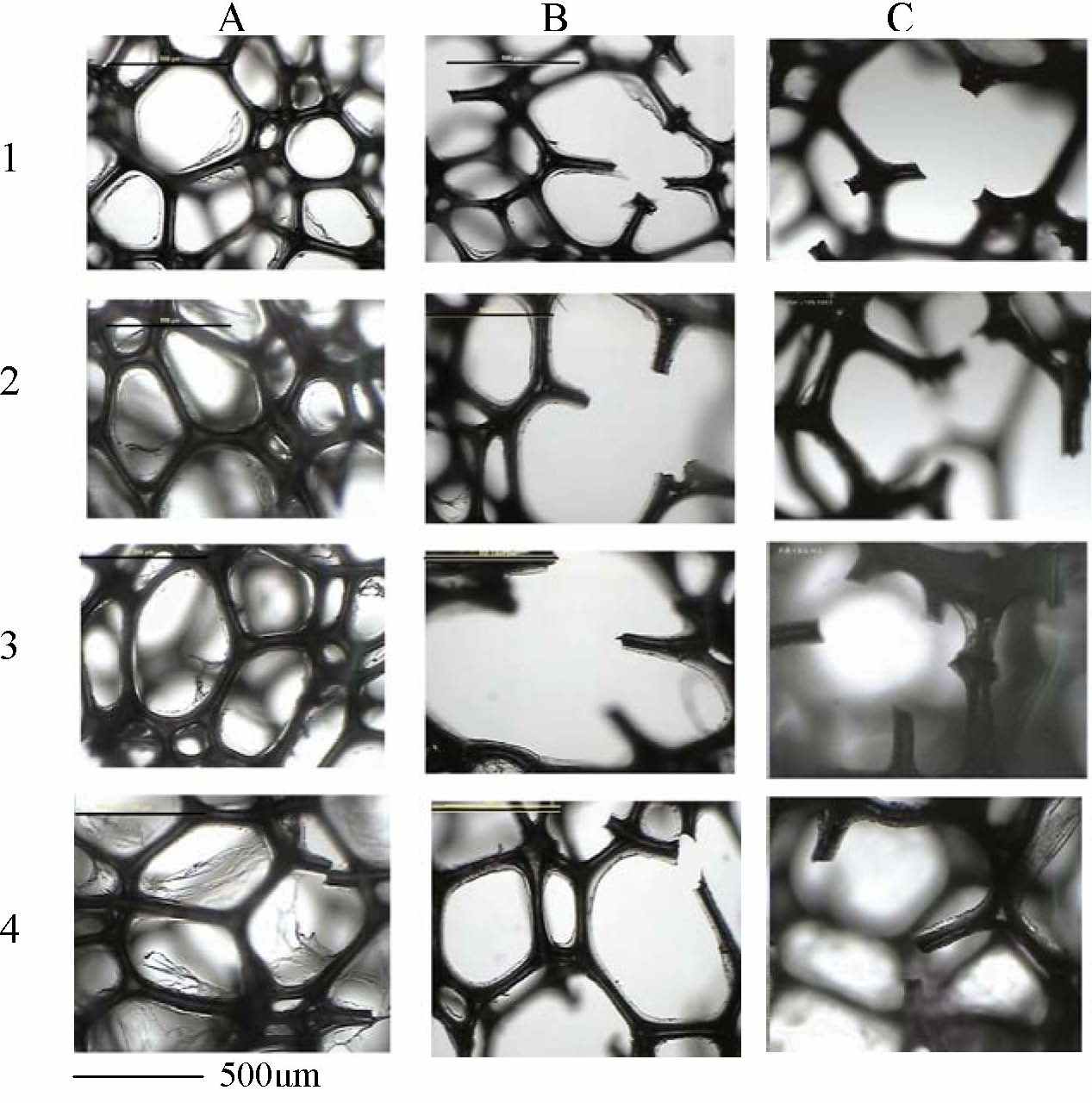
FIG. 2: Light micrographs of BIO-PUR
foams (1) ref. PUR, (2) with 10% HEC, (3) 10% AS, (4) 10%AC before
biodegradation (A) and after biodegradation by Thermophillus sp. (B) and Aureobasidium pullulans (C).
Biodegradability of these modified
materials is generally a function of the structure, the presence of
microbial population and the environment. Degradation activity (degree)
of BIO-PUR foams corresponded with the growth of the both Thermophillus and Aureobasidium culture.
The highest degree of the degradation was found in BIO-PUR modified by
10% of AS with about 4 times lower biomass in comparison with the
reference culture (without polyurethane). High degree of biodegradation
exhibited also the sample modified by 10% of HEC. On the other hand,
the BIO-PUR modified by 10% of AC was seemed to be the most stable
structure showing minimal growth changes. We presupposed that the
biodegradation of PUR foam modified by AC bio-polyol insoluble in
common polyether polyol proceeded from "outside" the polymer
determining very slow progress, while the biodegradation of BIOPU foams
modified by soluble AS and HEC proceeded from "inside" the PU network
implicating the higher rate of the degradation.
Possible migration of polymer
degradation products in the environment has to be monitored since
additional major pollutants that can be formed may endanger some of the
environmental components. Thus prepared BIO-PUR foams modified by AC,
AS, HEC and CMC were evaluated in respect to their ecotoxicity using
the alternative toxicity tests such as DAPHTOXKIT FTM MAGMA, ROTOXKIT F, ALGALTOXKIT FTM and THAMNOTOXKIT FTM.
BIO-PUR foams were heated with standard freshwater and toxicity of the
extracts was tested. The best results yielded THAMNOTOXKIT FTM toxicity
test showing the least toxic extract from the BIO-PUR foam modified by
AS bio-polyol, which exhibited even less toxicity than the original
ref. PUR.
Acknowledgement This work was supported
by the Ministry of Education, Youth and Physical Training of the Czech
Republic under the research project no. MSM 021630501.
References
- Oertel G.: Polyuretane handbook, 2nd ed., Hanser Publisher, 1993.
- Cunningham R. L., Carr M. E., Bagley E. B.: J. Appl. Polym. Sci., 1992, 44, 1447-1483.
- Rivera-Armenta J. L., Heinze Th., Mendoza-Martinez A. M.: Eur. Polym. J., 2004, 40, 2803-2812.
DETEKCE BIOMIMETICKÝCH SKUPIN NA
POLYLAKTIDOVÝCH POVRŠÍCH MODIFIKOVANÝCH FUNKCIONALIZOVANÝMI AMFIFILNÍMI
BLOKOVÝMI KOPOLYMERY
E. CHÁNOVÁ, Š. POPELKA, L. MACHOVÁ, V. PROKS, F. RYPÁČEK
Ústav makromolekulární chemie AV ČR, Heyrovského nám. 2, 162
06 Praha6. chanova(imc.cas.cz
Úvod
Biodegradabilní polymery na bázi polyesterů, jako např.
polylaktid
(PLA), jsou důležitými materiály v oblasti tkáňového inženýrství.
Jedním z podstatných faktorů, které ovlivňují chování buněk v kontaktu
s polymerem, je specifická distribuce bioaktivních molekul
podporujících buněčnou adhezi na povrchu biomateriálu. Takovými
molekulami mohou být peptidové sekvence odvozené ze struktury proteinu
mezibuněčné hmoty (např. sekvence -RGDS- fibronektinu) [1]. V naší
laboratoři jsou pro přípravu funkcionalizovaných biomimetických povrchů
biomateriálů na bázi PLA používány amfifilní blokové kopolymery
poly(laktid)u (PLA) a poly(ethylenoxid)u (PEO), které na konci PEO
bloku obsahují výše zmiňované peptidové sekvence, přičemž se
předpokládá jejich vliv na migraci, růst a diferenciaci buněk [2].
Pomocí modelových studií se snažíme získat představu o
topografii
povrchů, zvláště pak o distribuci a dostupnosti adhezních skupin (RGDS)
na površích, které je možné získat depozicí amfifilních kopolymerů na
povrch biomateriálu, případně jejich samoorganizací
("self-organization") na rozhraní biomateriál/voda. V této práci
používáme blokové kopolymery, které jsou na konci PEO bloku
funkcionalizovány biotinem. Sledováním specifické
interakce
biotinu s bílkovinou avidin [3] je možné studovat přístupnost funkčních
skupin na povrchu a vizualizací avidinu i jejich povrchové rozložení.
Experimentální část
Di-blokové kopolymery na bázi poly(DL-laktid)u (PDLLA) a
poly(ethylenoxid)u (PEO), s biotinem na konci PEO bloku (PDLLA-b-PEO-biotin)
a s rozdílnou délkou PEO bloků, byly připraveny polymerizací za
otevření kruhu (ring opening polymerization) DL-laktidu. Polymerizace
byla provedena v toluenu za použití α-biotinyl-ω-hydroxy-PEO jako
makroiniciátoru a 2-ethylhexanoátu cínu (II) (Sn(Oct)2)
jako
katalyzátoru. Biotinylovaný poly(ethylenoxid)ový makroiniciátor byl
připraven reakcí α-amino-ω-hydroxy-PEO (komerčně dostupný, NOF
Corporation) s p-nitrofenylovým esterem d-biotinu v dimethylformamidu
[4].
Neutrální kopolymery poly(DL-laktid)u a poly(ethylenoxid)u s
methoxy skupinou na konci PEO bloku (PDLLA-b-PEO-OMe)
a s odpovídající délkou jednotlivých bloků byly připraveny za podobných
podmínek, tj. polymerizací za otevření kruhu DL-laktidu, jako
makroiniciátor byl použit α-methoxy-ω-hydroxy-PEO připravený aniontovou
polymerizací ethylenoxidu iniciovanou směsí tvořenou monomethyletherem
triethylenglykolu a jeho draselným alkoholátem [5].
Připravené makroiniciátory a kopolymery byly charakterizovány 1H
NMR spektroskopií a GPC analýzou.
Připravené kopolymery byly rotačně naneseny z roztoků
obsahující
různý poměr neutrálního a biotinylovaného kopolymeru na podložku, která
byla tvořena homogenním filmem poly(L-laktid)u na rovném povrchu
(slída, sklo). Všechny rotačně nanesené kopolymery, včetně samotného
PLLA, byly inkubovány v roztoku avidinu (0,15mg/ml) v PBS (0,01M
fosfátový pufr, pH 7,5). Po inkubaci byly vzorky opakovaně opláchnuty
PBS a vodou a byly vysušeny v atmosféře dusíku.
Všechny typy povrchů byly charakterizovány jak před reakcí,
tak po
reakci s avidinem pomocí měření kontaktních úhlů a jejich topografie
byla analyzována pomocí mikroskopie atomárních sil (AFM) v tapping modu
na vzduchu.
Výsledky a diskuse
Využitím specifických interakcí biotinu s avidinem byla
sledována
dostupnost biotinových skupin na připravených površích vizualizací
povrchově vázaného avidinu (protein, MW 66KDa). Morfologie povrchů a
fázová separace PLA a PEO bloků byla posuzována vzhledem k molekulárním
parametrům použitých di-blokových kopolymerů a vzhledem k podmínkám
přípravy povrchů.
Získané výsledky prokázaly, že morfologie povrchů, které jsou
modifikovány amfifilními blokovými kopolymery, závisí na molekulárních
parametrech použitých kopolymerů. Tvorba odlišných struktur závisí na
schopnosti PEO řetězců krystalizovat a na fázové separaci PLA a PEO
bloků.
Předpokládaná nespecifická adsorpce avidinu, který zde
vystupuje i
jako protein obecně, na samotný PLLA povrch může být výrazně potlačena,
je-li PLLA povrch pokryt PLA-b-PEO kopolymery.
Výraznějšího efektu je pak dosahováno s PLA-b-PEO
kopolymery s delšími PEO bloky (viz Obr. 1A, D vs. Obr. 1B, E).
Přítomnost biotinové skupiny na modifikovaných površích s
potlačenou
proteinovou adsorpcí byla prokázána zvýšeným množstvím vázaného avidinu
(srovnej Obr. 1E a 1F).
Ačkoli nebylo možno, vzhledem k relativně velké drsnosti
polymerních
povrchů vůči velikosti molekuly avidinu, stanovit přesnou distribuci
biotinových skupin, selektivní vazba avidinu s biotinylovanými
kopolymery je známkou toho, že funkční biomimetické skupiny na površích
modifikovaných amfifilními PLA-b-PEO kopolymery jsou
na povrchu,
a proto mohou být dostupné pro interakce s jejich proteinovými
protějšky, například receptory na povrchu buňky.
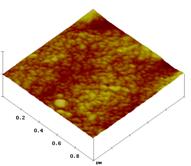
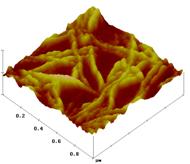




Obr. 1: Topografické AFM obrázky (1µm x
1µm, Z-scale 100nm) PLLA (A, D), PDLLA-b-PEO-OMe s
M(PEO)~15000 (B, E) a PDLLA-b-PEO-OMe/PDLLA-b-PEO-biotin
(hmotnostní poměr 9:1)
s M(PEO)~15000 (C, F). Obrázky (A), (B) a (C) byly pořízeny před
inkubací vzorků v roztoku avidinu, obrázky (D), (E) a (F) byly získány
po inkubaci s avidinem.
Za účelem vizualizace individuálních funkčních skupin bylo
využito
objemnější značky (avidin vázaný na nanočástice: NeutrAvidin-labeled
microspheres, Molecular Probes, velikost cca 0,04µm) za podmínek
podobných použití samotného avidinu, výjimkou byl nižší obsah
biotinylovaného kopolymeru v nanášené směsi (hmotnostní poměr PDLLA-b-PEO-OMe/PDLLA-b-PEO-biotin
199:1) (viz Obr. 2).
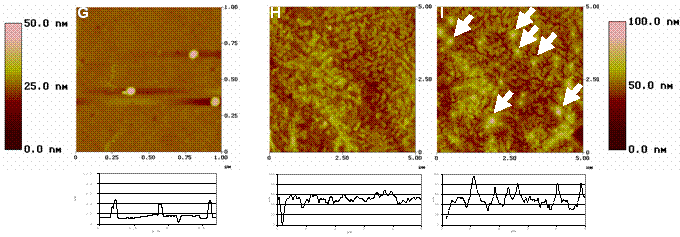
Obr. 2: Topografické AFM obrázky a jejich
reprezentativní
profily: nanočástice (mikrokuličky) na PLLA (G) (1µm x 1µm, Z-scale
50nm), PDLLA-b-PEO-OMe/PDLLA-b-PEO-biotin
(obsah PDLLA-b-PEO-biotin 0,5 hm.%)
(H) a ten samý povrch po inkubaci s kuličkami modifikovanými avidinem
(I) (oba 5µm x 5µm, Z-scale 100nm). Šipky: avidinem modifikované
kuličky navázané na biotinylovaný kopolymer.
Závěr
Selektivní vazba avidinu na povrchy modifikované blokovými
kopolymery, které nesou biotin jako skupinu zde reprezentující
peptidovou sekvenci RGDS, ukazuje, že koncové funkční skupiny
amfilfilních PLA-b-PEO blokových kopolymerů
deponovaných na PLA
povrchy jsou exponovány a dostupné pro interakce s jejich proteinovými
protějšky, např. povrchovými receptory buněk.
Využitím biotinu jako reprezentativní skupiny ve funkčních PLA-b-PEO
kopolymerech a značení avidinem v kombinaci s AFM představuje
praktickou metodu pro studie topografie povrchů funkcionalizovaných
biomateriálů.
Použitím objemnějších značek, jako jsou avidinem modifikované
mikrokuličky, umožňuje AFM detekci povrchové topografie individuálních
funkčních skupin, přičemž se předpokládá analogická distribuce i -RGDS-
peptidových sekvencí.
Reference:
[1] Hersel U., Dahmen C., Kessler H. Biomaterials
2003, 24, 4385.
[2] Bacakova L., Filova E., Kubies D., Machova L., Proks V.,
Lisa V., Rypacek F. J. Mater. Sci.-Mater. M. (in
press).
[3] Sakahra H., Saga T. Adv. Drug
Deliver. Rev. 1999, 37, 89.
[4] Salem A.K., Cannizzaro S.M., Davies M.C., Tendler S.J.B.,
Roberts C.J., Williams P.M., Shakesheff K.M. Biomacromolecules
2001, 2, 575.
[5] Popelka S., Machova L., Rypacek F., Spirkova M., Stepanek
M., Matejicek P., Prochazka K. Collect. Czech. Chem. Commun.
2005, 70, 1811.
Poděkování: Práce byla uskutečněna s
podporou Grantové
agentury Akademie věd České republiky (grant číslo: A4050202), Centrem
buněčné terapie a tkáňových náhrad (Ministerstvo školství ČR, grant
číslo: 1M0021620803) a 6FP EU "EXPERTISSUES" (NMP3-500283-2).
NANOKOMPOZITY NA BÁZI SILIKONOVÉHO KAUČUKU
V. RABOVÁ, P. HRON
Ústav polymerů, Vysoká škola chemicko-technologická v Praze,
Technická 1903, Praha 6‑Dejvice, 166 28, Česká republika (rabova.v(centrum.cz,
hronp(vscht.cz)
Chemie v nanometrickém měřítku jako poddisciplína
materiálových a
chemických věd se zabývá vývojem metod pro přípravou nanoskopických
materiálů uvnitř pórů a vrstev mikroporézních materiálů. Typickým
příkladem nanotechnologie je problematika polymerních nanokompozitů,
která v posledních několika letech zaujala pozornost vědců,
zabývajících se vylepšováním vlastností polymerů, respektive
polymerních kompozitů.
Polymerní nanokompozit je tvořen polymerní matricí, ve které
je
dispergováno plnivo, vyznačující se dále uvedenou strukturou. Vzájemnou
interakcí vznikají tři základní typy struktur:
- Interkalovaný kompozit
- Exfoliovaný kompozit
- Delaminovaný kompozit
Interkalace je zabudování atomů, iontů, resp.molekul hosta do
vhodné, nejlépe vrstevnaté hostitelské struktury. Při exfoliaci jílu
jsou silikátové vrstvy hostitelské struktury od sebe natolik vzdálené,
že již nelze mluvit o pravidelné vrstevnaté struktuře, ve které se
střídají organické vrstvy se silikátovými vrstvami.
V současné době je velká pozornost kladena na jílovitá
plniva,
která jsou řazena mezi vrstevnaté materiály s vrstvami širokými 1 nm,
poutanými Van der Walsovými vazbami. Typickým zástupcem je
montmorillonit - hlinitokřemičitý hydrofilní materiál, který vzniká
zvětráváním čedičových půd a hornin a sopečných popelů. Povrch vrstvy
je tvořen hlavně tetraedrálním křemíkem, centrální rovina vrstvy
obsahuje oktaedrálně koordinovaný Al3+, často je
možná nestechiometrická substituce, kde je Al3+
nahrazen Mg2+, případně Fe3+ 45.
Mg2+
substituce zanechává vložený záporný náboj v jílu, který musí být
neutralizován kationtem na povrchu. Většinou je to kation typu Na+,
užit však může být i tetrasubstituovaný amoniový ion, který slouží jako
iontově vázaný organický modifikátor. Touto úpravou se sníží
hydrofilita montmorillonitu a tím vzrůstá adheze hydrofobních polymerů
a plniva. Struktura montmorillonitu je patrná z obrázku 1 (červeně -
Si, růžově - Al, modře - O, šedomodře - OH, zeleně - Na).
Obr. 1
Příprava nanokompozitů na bázi kapalného silikonového kaučuku
byla
prováděna polymerizací cyklických oligomerů in situ v přítomnosti
termolabilního iniciátoru (TMAH) s užitím různě povrchově
modifikovaných montmorillonitů, případně bez přítomnosti termolabilního
iniciátoru, kde by samotná amoniová sloučenina měla schopnost iniciovat
daný systém (počáteční homogenizace byla prováděna ve skleněné baňce s
míchadlem a v ultrazvukové lázni). Další typy směsí byly připravovány
homogenizací silikonového kaučuku Lukopren N1000 (příp. Silopren K
10000) a nanoplniva (příp. tradičního neaktivního nebo aktivního
plniva) na třecím tříválci a homogenizací ultrazvukovým míchadlem.
Použitím různým způsobem povrchově modifikovaných
montmorillonitů
byly připraveny směsi značně se lišící hodnotami zdánlivých viskozit.
Při homogenizaci OKT s nanoplnivem v baňce s míchadlem a v ultrazvukové
lázni dosáhla zdánlivá viskozita hodnoty o jeden řád vyšší než u směsí
připravených homogenizací Lukoprenu N1000 a nanoplniva na třecím
tříválci.
U připravených směsí byla měřena závislost zdánlivé viskozity
na
smykové rychlosti a bylo potvrzeno pseudoplastické chování připravených
směsí, tj. se vzrůstající smykovou rychlostí klesá zdánlivá viskozita.
Po 24 hodinách odležení směsí došlo v některých případech k vzrůstu
nebo poklesu zdánlivé viskozity, což lze vysvětlit jako následek dvou
probíhajících dějů - sedimentace a mezimolekulárních interakcí. Jsou-li
interakce polymer - plnivo intenzivní, je potlačena sedimentace a
zdánlivá viskozita během odležení vzrůstá. Nedochází-li k interakcím
nebo jsou-li malé, dochází k usazování plniva (před měřením nebyly
směsi záměrně z důvodu porušení případných struktur zamíchány).
Směsi byly vulkanizovány za normální teploty pomocí
katalyzátoru C21
v množství 4 dss a z vulkanizátů pak byla vysekávána zkušební tělíska k
měření mechanických vlastností.
Z mechanických vlastností byla pozornost věnována především
pevnosti
a tažnosti. Hodnoty pevnosti v tahu jsou závislé na způsobu přípravy
směsí - probíhá-li příprava polymerizací OKT v přítomnosti nanoplniva a
za iniciace TMAHem, pevnost v tahu nabývá vyšších hodnot než v případě
směsí připravených homogenizací v ultrazvukové lázni a na třecím
tříválci.
V případě pouze jednoho typu plniva nebylo nutno při
polymerizaci v
baňce s míchadlem použit termolabilní iniciátor, protože z předchozích
experimentů bylo zjištěno, že amoniová sloučenina, použitá k povrchové
úpravě vrstevnatého silikátu, působí jako iniciátor polymerizace. Tato
domněnka byla potvrzena u směsi připravené polymerizací
oktamethylcyklotetrasiloxanu a nanoplniva (10 dsk), kde mechanická
pevnost dosáhla maximální hodnoty, což je hodnota přibližně stejná jako
u směsi Lukoprenu N 1000 a trojnásobně vyšší koncentraci křemeliny.
Měřením mechanických vlastností byly potvrzeny výsledky
zveřejněných
prací, které prezentují zvýšení hodnot pevnosti v tahu užitím nanoplniv
při nižším stupni plnění oproti tradičně užívaným plnivům užívaných v
technologii kapalných silikonových kaučuků.
Ke studiu termické stability byla užita metoda
termogravimetrické
analýzy, která umožnila stanovit teploty rozkladu nízkomolekulárních
podílů, TMAHu, povrchové úpravy silikátu a siloxanové složky.
Cílem práce bylo připravit nanokompozity na bázi kapalného
silikonového kaučuku a zjistit, jakým způsobem budou ovlivněny hodnoty
zdánlivých viskozit a mechanických vlastností při různém stupni plnění
a užitím různě povrchově modifikovaných montmorillonitů, případně jak
se tyto hodnoty budou lišit od hodnot získaných u směsí připravených z
tradičních aktivních a neaktivních plniv užívaných u licích
silikonových kaučuků.
ŠTÚDIUM KOMPOZITOV NA BÁZE MONTMORILLONITU S
BIODEGRADOVATEľNOU MATRICOU
I. JANIGOVÁa, Z. NÓGELLOVÁa,
I. CHODÁKa, F. LEDNICKÝb
aÚstav polymérov SAV, Dúbravská cesta
9, 842 36 Bratislava, Slovenská republika (upoljani(savba.sk)
bÚstav makromolekulární chemie AV ČR,
Heyrovského nám. 2, 162 06 Praha 6, Česká republika
Je známe, že prítomnosť plniva v polymérnej matrici ovplyvňuje
vlstnosti kompozitných materiálov. Montmorillonit (MMT) ako plnivo v
polymérnej matrici je vhodný vzhľadom na doštičkovú štruktúru,
dostupnosť v prírode a nízku cenu. Modifikáciou vrstevnatého MMT alebo
matrice môže dochádzať k interkalácii a/alebo exfoliacii vrstiev plniva
pri príprave za vzniku nanokompozitov, pričom sa získajú materiály so
zlepšenými vlastnosťami (mechanické, termické) už pri malom obsahu
plniva. V posledných rokoch dôležitú oblasť záujmu predstavujú
kompozity s biodegradovateľnou matricou, pri ktorých obsah
nebiodegradovateľného plniva nesmie prekročiť 5 hm.%, aby sa zachovala
biodegradabilita materiálu.
Predložená práca je zameraná na štúdium morfológie a
mechanických
vlastností kompozitov s biodegradovateľnou matricou s možnosťou vzniku
nanokompozitov pri príprave konvenčným miešaním v tavenine. Zvolili sa
dva typy biodegradovateľných matríc - polykaprolaktón (PCL) a zmes
PCL/termoplastický škrob (≈ 50/50 hmot.%, MaterBi). Ako plnivo sa
aplikovali štyri typy MMT, ktoré sa líšili veľkosťou častíc a
modifikáciou. (Nanofil 15 < J10 < BJ40 < Kunipia).
Ako jediný
čiastočne interkalovaný MMT sa použil Nanofil 15.
Morfológia kompozitov pripravených konvenčným miešaním v
tavenine
sa sledovala metódami elektrónovej miroskopie SEM a TEM. Mikrografy SEM
v prípade kompozitov s Nanofilom 15 v obidvoch polymérnych matriciach
ukázali čiastočne exfoliované vrstvy plniva v celom objeme matrice
(Obr. 1a, 2a) bez prítomnosti pôvodných častíc plniva. TEM mikrografy
tých istých materiálov nielen potvrdili prítomnosť čiastočne
exfoliovaných vrstiev Nanofilu 15 ale aj existenciu exfoliovaných
vrstvičiek plniva (Obr. 1b, 2b). Na základe mikroskopických obrázkov je
možné konštatovať, že v prípade interkalovaného plniva Nanofilu 15 sa
pripravil konvenčným miešaním v tavenine v Brabendri nanokompozit.
V kompozitoch plnených J10 a BJ40 napriek tomu, že sa
zachovala
väčšina plniva vo forme častíc, na mikroskopických obrázkoch sa
pozorovali aj čiastočne exfoliované vrstvy MMT v obidvoch typoch
matríc, nie je však možné hovoriť o nanokompozite.
a b
Obr.1: SEM (a) a TEM (b) mikrografy vzorky PCL/Nanofil 15 (3
hm.%).
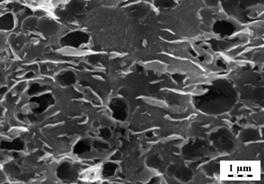
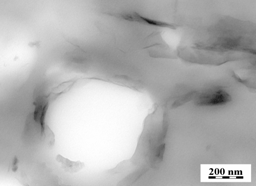
a b
Obr.2.: SEM (a) a TEM (b) mikrografy vzorky MaterBi/Nanofil 15
(3 hm. %).
Hoci SEM pozorovania naznačovali i v prípade Kunipie ako
plniva s
najväčšími pôvodnými časticami (> 40 µm) možnosť čiastočnej
exfoliacie, na TEM mikrografoch sa objavovali len ojedinelé čiastočne
interkalované a/alebo čiastočne exfoliované častice.
Výsledky získané z mechanických vlastností korelovali s
mikroskopickými pozorovaniami pri matrici PCL, kde pre vzorku s
Nanofilom 15 ako plnivom sa dosiahlo najvýraznejšie zlepšenie
mechanických vlastností (E, ε, б). Na druhej strane najhoršie
mechanické vlastnosti sa zaznamenali pre vzorku s MMT Kunipia. Pri
kompozitoch s matricou MaterBi je situácia komplikovanejšia
pravdepodobne kvôli nižšej adhézii v matrici, čo naznačili SEM obrázky,
kde
na lomových plochách sa pozorovali diery po zrnkách škrobu.
Tento
efekt sa prejavil už na výraznom poklese hodnôt predĺženia pri
pretrhnutí v samotnej zmesi bez prítomnosti plniva v porovnaní s
hodnotami pre matricu PCL. Kompozity s matricou MaterBi zaznamenali
rast hlavne pri hodnotách Youngovho modulu.
Poďakovanie
Autori ďakujú za finančnú podporu "Agentúre pre podporu vedy"
(Projekt č. APVV-51-050505)
štruktúra a elektrická vodivosť
Polypropylén/montmorilLonit/polypyrolových kompozitov
M. OMASTOVÁa, M. MRAVČÁKOVÁa,
J. PROKEŠb
a Ústav polymérov, Slovenská academia
vied, Dúbravská cesta 9, 842 36 Bratislava, Slovensko
(upolmaom(savba.sk)
b Karlova Univerzita v Prahe, Fakulta
matematiky a fyziky, Katedra makromolekulárni fyziky,
Ke Karlovu 5, 121 16 Praha, Česká republika
Použitie polymérov je možné rozšíriť prídavkom plnív.
Prítomnosť
nanočastíc vrstevnatých silikátov, ako je napr. montmorilonit (MMT), v
polymérnej matrici môže viesť k výraznému zlepšeniu fyzikálnych a
chemických vlastností polymérov, vo väčšej miere ako prítomnosť
mikroplniva, vzhľadom na jeho väčší merný povrch a súčasne väčší pomer
dlhšieho rozmeru ku kratšiemu. Vlastnosti týchto nanokompozitov výrazne
ovplyvňuje distribúcia MMT nanovrstiev v polymérnej matrici.
Špeciálnu triedu polymérov tvoria vodivé polyméry, medzi
ktoré
patria polypyrol (PPy), polyanilín, polytiofén a iné. Tieto polyméry sú
predmetom širokého výskumu, súvisiaceho s možnosťou ich aplikácie v
rôznych oblastiach priemyslu, ako elektronické zariadenia, spínače
citlivé na tlak, dôležité strategické materiály, ako materiály pre
elektromagnetické tienenie, materiály na rozptýlenie statického náboja
a iné. Ich ťažká spracovateľnosť a v určitých prípadoch nevyhovujúce
mechanické vlastností sú dôvodom, že sa skúmajú v kombinácii s inými
materiálmi. Röntgenová fotoelektrónová spektroskopia MMT/PPy
nanokompozitov, ktoré boli pripravené chemickou oxidačnou
polymerizáciou pyrolu v prítomnosti exfoliovaného MMT a mali veľmi
nízku vodivosť ukázala, že povrch MMT je málo pokrytý PPy, keďže pyrol
prednostne interkaluje medzi vrstvy sodného typu MMT [1]. Na
súvislejšie pokrytie povrchu tohto typu MMT je potrebné množstvo pyrolu
zodpovedajúce viac ako 10 hm. % [2], čo sa prejaví následným rastom
vodivosti kompozitu. Ak polymerizácia prebieha na povrchu organofilného
MMT už nízke množstvo pyrolu v reakčnej zmesi vedie k vzniku vodivého
kompozitu.
Jedným zo spôsobov prípravy vodivých nanokompozitov je
modifikácia
systému polymérna matrica - anorganické nanoplnivo vodivým polymérom.
Vytvorenie vodivej siete, ktorá prestupuje nevodivou polymérnou
matricou, a zároveň určitým spôsobom obaľuje nanočastice plniva, vedie
k získaniu vodivých nanokompozitov. Oblasťou nášho záujmu sú
nanokompozity obsahujúce polypropylén (PP) ako polymérnu matricu,
anorganické plnivo - MMT sodného typu (MMT BJ) a polypyrol ako vodivú
zložku, ktoré boli pripravené v dvoch reakčných roztokoch a spracované
rozličnými metódami. Na základe získaných výsledkov pre pripravené
kompozity sme študovali vplyv obsahu PPy, MMT, ako aj spôsobu prípravy
a spracovania na ich výsledné vlastnosti. Štruktúra kompozitov
obahujúcich MMT bola analyzovaná širokouhlovou röntgenovou difrakciou
(WAXS), rastrovacou elektrónovou mikroskopiou (SEM) a transmisnou
elektrónovou mikroskopiou (TEM). Na zhodnotenie stavu disperzie MMT v
kompozitoch sme použili reológiu.
Trojzložkové polypropylén/montmorilonit/polypyrolové
(PP/MMT/PPy)
kompozity sme pripravili chemickou oxidačnou polymerizáciou pyrolu v
suspenzii rozdispergovaných nanočastíc MMT a PP častíc. Pre porovnanie
morfólogie a vlastností boli pripravené dvojzložkové PP/PPy a PP/MMT
kompozity. Všetky kompozity sme pripravili dvoma spôsobmi: PP sme pred
polymerizáciou pyrolu rozdispergovali v roztoku 1.) voda/metanol (V/V =
1/1) alebo 2.) vo vodnom roztoku surfaktantu,kyseliny
dodecylbenzénsulfónovej (DBSA, n(Py)/n(DBSA) = 5/1). K vzniknutej
suspenzii sme pridali MMT v hmotnostnom pomere wMMT/wPP
= 5/100. Z dôvodu získania exfoliovanej štruktúry MMT sme počas 10 min.
na suspenziu aplikovali ultrazvuk. Následne počas miešania sme pridali
roztok oxidačného činidla FeCl3 vo vode (n(FeCl3)/n(Py)
= 2,3). Posledným krokom prípravy kompozitov bolo postupné pridanie
pyrolu (1 až 16,7 hm.%). Práškové kompozity získané po filtrovaní a
sušení boli následne spracované dvoma spôsobmi: 1.) priamym lisovaním
pri 180°C alebo 2.) miešaním v tavenine pri 190°C a následným
lisovaním.
Vlastnosti PP/PPy a PP/MMT/PPy kompozitov sú vo veľkej miere
ovplyvnené metódou a podmienkami polymerizácie pyrolu, množstvom
vzniknutého PPy, ako aj spôsobom spracovania vzoriek [3]. V PP/MMT/PPy
kompozitoch, ktoré boli spracované priamym lisovaním, ostala zachovaná
fyzikálna vodivá sieť MMT/PPy častíc a PPy, čo sa prejavilo vysokými
vodivosťami. Kompozit obsahujúci 4,8 % PPy v kompozite mal vodivosť asi
10-5 S cm-1
[2]. Metódou
WAXS bolo potvrdené pokrytie MMT nanočastíc v PP/MMT/PPy
nanokompozitoch polypyrolom, pričom DBSA prispela k homogénnejšiemu
rozptýleniu MMT vrstiev a zvýšila vodivosti PP/PPy a PP/MMT/PPy
kompozitov pri porovnaní s kompozitmi pripravenými vo vodnom roztoku
metanolu modifikovaných rovnakým množstvom pyrolu. Výsledky TEM
potvrdili interkaláciu a čiastočnú exfoliáciu MMT v PP/MMT/PPy
kompozitoch.
Pre binárne PP/PPy kompozity sa spracovaním homogenizáciou v
tavenine pozorovala deštrukcia vodivej siete, čo sa prejavilo nízkou
vodivosťou a komplexnou viskozitou. Pre PP/MMT/PPy kompozity pripravené
použitím DBSA, obsahujúce nad 9,1 hm.% PPy boli namerané vodivosti v
rozsahu od 10-5 do 10-3
S cm-1,
čo naznačuje že prítomnosť nanoplniva stabilizuje vodivú sieť. Štúdiom
pripravených PP/MMT/PPy kompozitov s obsahom 13,0 hm.% PPy a rôznym
obsahom MMT sme zistili, že že už 1 hm.% MMT v PP/MMT/PPy kompozite je
postačúce na stabilizáciu vodivej siete počas spracovania
homogenizáciou v tavenine.
Pripravili sme nový typ kompozitného materiálu PP/MMT/PPy,
ktorý pri
obsahu vodivej zložky vyššom ako 9,1 hm.% PPy dosahuje hodnotu
elektrickej vodivosti, ktorá ho zaraďuje medzi antistatické materiály.
Tento kompozit by mohol nájsť využitie ako materiál na
elektromagnetické radiačné tienenie.
Poďakovanie
Táto práca bola finančne podporená Vedeckou
a grantovou
agentúrou Ministerstva školstva SR a Slovenskej akadémie vied (VEGA
2/4024/04) a Ministerstvom školstva, mládeže a športu Českej republiky
MSM 0021620834).
Literatúra
- K. Boukerma, J.-Y. Piquemal, M. M. Chehimi, M. Mravčáková, M.
Omastová, P. Beaunier, Polymer, 47 (2006) 569.
- M. Mravčáková, M. Omastová, P. Pötschke, A. Pozsgay, B.
Pukánszky, J. Pionteck, Polym. Adv. Technol., v tlači.
- M. Omastová, M. Mravčáková, I. Chodák, J. Pionteck, L.
Häussler, Polym. Eng. Sci, 46 (2006) 1070.
INVESTIGATION OF PROPERTIES OF
PRESSURE-SENSITIVE ADHESIVES WITH ELECTRIC CONDUCTIVITY
Š. FLORIÁN, I. NOVÁK
Polymer Institute, Slovak Academy of Sciences, Dúbravská cesta
9,
842 36 Bratislava, Slovakia
E-mail: upolflor(savba.sk
The conductivity of electro-conductive adhesives in the
direction of
pressure action is to a certain extent dependent on the magnitude of
pressure applied. Electrically conductive pressure-sensitive adhesives
(PSA) are used mainly for production of electrically conductive tapes
consisting of backing and adhesive that must combine typical PSA
characteristics with excellent electrical properties 2, 3.
Electrical grade PSA are designed and manufactured using materials that
are physically and chemically stable in the presence of humidity and
electrical stress. In the contribution electrical conductivity of
acrylic-based PSA filled with carbonaceous filler has been studied.
Thus the percolation concentration can be reached if the content of the
filler in PSA composite exceeds 10 wt. % for carbon black or 30 wt. %
for graphite. Because of intensive decrease in strength of adhesive
joint and simultaneous satisfactory electrical parameters it is
necessary to use the concentration of carbonaceous filler that
corresponds to begin of stabilization of electrical conductivity after
exceeding the percolation concentration of filler in PSA composite.
The authors are grateful to Slovak grant
agency VEGA (grant No. 2/4042/04) for the financial support of this
research.
SURFACE AND ADHESIVE PROPERTIES OF
POLYETHYLENE MODIFIED BY ELECTRICAL DISCHARGE PLASMA
Igor Novák* , Marián Števiar, Ivan Chodák,
Igor Krupa, T. Nedelčev, M. Špírková1, M. Chehimi2,
A. Kleinová, O. Šolcová3
Ústav polymérov SAV, Dúbravska cesta 9, 842
36 Bratislava, Slovakia
1 Institute of
Macromolecular Chemistry,
Academy of Sciences of the Czech Republic, Heyrovskeho nám. 2, 162 06
Praha 6, Czech Republic
2 Interfaces,
Traitements, Organisation et Dynamique des Systèmes (ITODYS), Université Paris 7-Denis Diderot, 1 rue Guy
de la Brosse, 75005 Paris, France
3 Ústav chemických
procesu, Academy of Sciences of the Czech Republic, Rozvojová 135, 165
02 Praha 6, Czech Republic
E-mail: upolnovi(savba.sk
The low-density polyethylene (LDPE) was modified by barrier
discharge plasma at atmospheric pressure to improve adhesive properties
of polyethylene to improve surface properties, and to form a new
surface containing polar functional groups incoming to reactions with
some reactive compounds. The contact angles for characterization of the
surface properties of polymer were measured using SEE [Surface Energy
Evaluation] system and strengths of adhesive joints in peeling of the
modified polymer were tested on dynamometer Instron. The topography of
modified polyethylene was studied using Atomic Force Microscopy (AFM),
and changes in chemical structure were analyzed with X-ray
Photoelectron Spectroscopy (XPS). The significant increase of the
surface energy and its polar component of LDPE modified by surface
barrier discharge (SBD) plasma in oxygen or nitrogen plasma at
atmospheric pressure were confirmed. This increase significant even of
for short times of modification of polymer by plasma, and was further
increased for longer times of modification. The XPS analysis of
modified LDPE showed an increase in O/C atomic ratio from 0.011 to 0.16
after plasma modification of LDPE. This dramatic change affected the
atomic ratios as well as the C1s structure of the C1s peak. The
formation of a variety of nitrogen- and oxygen-containing groups, i.e.
C-N, C-O, C=O, O-C=O and N-C=O has been detected. AFM measurements of
LDPE modified by SBD plasma showed no significant differences in height
profiles, but demonstrate some melting marks on the surface of the
polymer.
The authors are grateful to the Slovak grant agency VEGA
(grant No. 2/4042/04) for the financial support of this research.
Modifikace polyamidu 6 aniontovou polymerací
J. RODA, J. BROŽEK
Ústav polymerů, Vysoká škola chemicko-technologická v Praze,
Technická 5, 166 28 Praha 6, Česká republika (Jan.Roda(vscht.cz)
Polyamid 6 - poly(e-kaprolaktam) (PA 6) patří mezi plasty s
vynikajícími fyzikálně-mechanickými a chemickými vlastnostmi. Využívá
se především v textilním průmyslu pro přípravu kvalitních vláken, avšak
skoro jedna třetina jeho produkce nachází uplatnění v oblasti
konstrukčních plastů. V příštích pěti letech se očekává min. 5%ní
nárůst spotřeby v této oblasti. Proto je třeba se zamýšlet nad
možnostmi další modifikace vlastností PA 6, jejich úpravou a
vylepšováním.
Aniontová polymerace laktamů, respektive e-kaprolaktamu (KL),
není
zdrojem většiny polymeru (PA 6) připraveného průmyslově, ale má
nezastupitelné místo při polymeračním odlévání a RIM technologii, tj.
přípravě i komplikovaných výrobků s relativně vysokou přidanou
hodnotou. Právě aniontová polymerace resp. aniontový mechanismus této
polymerace, umožňuje poměrně snadno modifikovat vlastnosti PA 6.
PA 6 má poměrně nízkou vrubovou houževnatost při teplotách
pod
bodem mrazu, kterou lze zlepšit syntézou dvoufázového systému a to
zabudováním elastomerní fáze do polyamidové matrice.
Aniontová polymerace využívá postupu, kdy se na koncích
prekurzorů elastických segmentů (in situ nebo extra
situ)
vytvoří neiontová růstová centra pro tvorbu polyamidových bloků. V
další fázi procesu po přídavku aniontového iniciátoru (nejčastěji sodné
soli e-kaprolaktamu - KLNa) vznikne blokový kopolymer PA 6 - elastomer
s vhodnou fázovou separací.
Polymeračním odléváním se podařilo připravit blokové
kopolymery PA
6 - blok - polybutadien. Polymerační procedura je poměrně jednoduchá.
Buď se v tavenině KL rozpustí a,w-dihydroxy-polybutadien (DHPBD) a
aromatický isokyanát a pak se polymerace ve formě vyvolá přídavkem
KLNa, nebo předem zreaguje a,w-dihydroxypolybutadien, diisokyanát a KL
a takto připravený makroaktivátor ve směsi s taveninou KL a KLNa
poskytne houževnatý materiál.
Bylo prokázáno, že DHPBD je zcela chemicky vázán ve struktuře
materiálu. Obsahem tohoto elastomeru lze regulovat morfologii a tak
houževnatost materiálu, která roste řádově oproti čistému PA 6. Nárůst
houževnatosti je pochopitelně doprovázen poklesem pevnosti a modulu,
rozumný kompromis vlastností lze poměrně snadno dosáhnout.
Další možností modifikace vlastností PA 6 je kopolymerace KL
s
dalšími laktamy, nejdostupnější je průmyslově vyráběný w-laurolaktam
(LL). Aniontová kopolymerace KL s LL iniciovaná alkalickými solemi
laktamů poskytuje statistické kopolymery. Při studiu iniciační aktivity
e-kaprolaktam-magnesiumbromidu (KLMgBr) bylo zjištěno, že při
polymeračních teplotách "hluboko" pod teplotou tání PA 6, kdy
polymerační proces má heterogenní charakter, vzniká materiál
(kopolymer) s dvěma endothemy tání.
Selektivní extrakcí a NMR analýzou bylo zjištěno, že materiál
je
tvořen statistickým kopolymerem KL a LL a blokovým kopolymerem tvořeným
bloky KL jednotek (tj. PA 6) a bloky statistického kopolymeru.
V případě iniciace kopolymerace KLMgBr v teplotním intervalu
150-180 °C (platí pro ekvimolární směs KL a LL) bylo zjištěno, že na
počátku polymerace se přednostně spotřebovává KL
(v podstatě se tvoří PA 6). LL se zřetelně zabudovává do struktury
kopolymeru až po spotřebě většiny KL dvěma reakcemi - propagací a
transamidačními výměnnými reakcemi. Tento jev nebyl pozorován za
stejných experimentálních podmínek při iniciaci kopolymerace KLNa,
ovšem při polymerační teplotě 120 °C byl isolován materiál tvořený dle
DSC také PA 6 a statistickým kopolymerem KL a LL. Takže tvorba
homopolymeru, statistického a blokového kopolymeru není výlučné
vlastností KLMgBr, ale je obecným rysem aniontové kopolymerace KL a LL.
Tvorba a zastoupení jednotlivých složek v materiálu je řízeno kineticky
v závislosti na typu a složení iniciačního systému a polymerační
teplotě. Například při polymeračním odlévání směsi KL a LL iniciované
KLMgBr se tvoří statistický kopolymer "vyztužený" PA 6, takže má lepší
mechanické vlastnosti než materiál připravený s KLNa jako iniciátorem.
Materiál také překvapivě obsahuje dvě rozdílné krystalické fáze.
KLMgBr je také jediný účinný aniontový iniciátor pro zajímavou
kopolymeraci KL s e-kapro-laktonem (KLO). Mechanismus propagace obou
cyklů je zcela odlišný. Výsledkem kopolymerace je statistický kopolymer
s kvantitativně zabudovaným KLO.
Předpokládaný mechanismus je formálně obdobný kopolymeraci
KL/LL.
Nejprve se vytváří velmi rychle (dokonce již při přípravě polymerační
násady) poly(e-kaprolakton) (PKLO), jeho esterové skupiny pak reakcí s
laktamovým aniontem vytváří N-acyllaktamové struktury, na kterých dále
probíhá polymerace KL. Za aktivní účasti výměnných (randomizačních)
reakcí, hlavně transacylačních se rychle tvoří statistický kopolymer.
Tento proces lze považovat za jednoduchou jednostupňovou přípravu
polyesteramidů (PEA). Zabudované KLO jednotky v kopolymeru zlepšují
jeho houževnatost
a upravují další vlastnosti.
Kopolymery - PEA mají nižší krystalinitu než PA 6 a společně s
obsahem relativně labilních esterových vazeb by se mohly zařadit mezi
biodegradovatelné materiály. Biodegradace PEA s rozdílným obsahem KLO
jednotek byla testována abiotickou hydrolýzou, kompostovacím testem
a účinkem ligninolytických hub.
Tato práce byla řešena jako součást
výzkumného záměru MSM 6046137302.
"PHASE CHANGE MATERIALS (PCM) " NA BÁZE
POLYETYLÉNU A PARAFÍNU
G. MIKOVÁ, I. KRUPA, I. CHODÁK
Ústav Polymérov, Slovenská Akadémia Vied, Dúbravská cesta 9,
842 36
Bratislava, Slovenská Republika (gizela.mikova(savba.sk,
http://www.polymer.sav.sk/ )
Potenciál zmesí polyolefínov s parafínovými voskami tkvie v
schopnosti voskov prechádzať fázovou premenou, pričom polyolefínová
matrica zabezpečuje udržanie kompaktného tvaru materiálu počas tejto
zmeny, a teda slúži ako skelet zabraňujúci úniku parafínu z materiálu.
Na základe uvedenej schopnosti sa tieto látky zaraďujú so skupiny tzv. Phase
Change Materials
(PCM), o ktorých štúdium je záujem najmä z hľadiska ich využitia ako
materiálov schopných uchovávať tepelnú energiu, respektíve poskytovať
ochranu pre náhlej zmene teploty okolia [1-3]. Tepelná energia okolia
sa spotrebuje na topenie vosku, pričom teplota materiálu ostáva
viac-menej konštantná. Naopak, pri poklese teploty parafín kryštalizuje
za súčasného uvoľňovania kryštalizačného tepla do okolia. Z komerčného
hľadiska sú uvedené systémy zaujímavé pri výrobe solárnych panelov,
alebo ako prímesi do konštrukčných materiálov. Ideálny PCM použitý na
uchovávanie energie by mal byť o.i. štruktúrne stabilný, vysoko tepelne
vodivý, chemicky inertný, netoxický, nehorľavý a cenovo dostupný. V
práci sme sa zamerali na prípravu zmesí nízkohustotného polyetylénu
(LDPE) s nízkomolekulovým, mäkkým parafínom (parafín S), ako aj
vysokomolekulovým, tvrdým Fisher - Tropschovým parafínom (parfín FT).
Parafín S vykazuje na DSC zázname dva oddelené, intenzívne
píky.
Prvý pík pri 41 °C zodpovedá prechodu medzi dvomi kryštalickými fázami,
druhý pík odpovedá teplote topenia kryštalitov pri 57 °C. Parafín FT má
prechod medzi kryštalickými fázami posunutý k teplote 72 °C a teplota
topenia kryštalitov leží pri 95 °C. V tomto prípade však píky nie su
tak ostro separované ako v pripade parafínu S. V zmesiach s
nízkohustotným polyetylénom sa oba parafíny chovajú rôzne. Parafín S sa
v zmesiach topí, respektíve kryštalizuje viacmenej neávisle, aj keď
čiastočne dochádza k posunom píkov topenia a kryštalizácie tak u
parafínu S ako aj u LDPE. V prípade parafínu FT však dochádza k
výraznej kokryštalizácii s polyetylénovou matricou. Na lepšiu
miešateľnosť parafínu FT s matricou poukazujú aj SEM snímky, kde vidno
že parafín S sa na rozdiel od parafínu FT viditeľne fázovo separuje.
Dynamicko mechanická termická analýza bola okrem stanovenia
dynamicko mechanických vlastností využitá aj na merania mechanickej
pevnosti a schopnosti deformácie materiálov pri určitých teplotách a
tiež na sledovanie teplotnej rozťažnosti a rozmerovej stálosti počas
cyklického termického namáhania vzoriek. Cyklovanie od -20 do 80 °C
bolo zvolené s cieľom simulovať podmienky zaujímavé pre materiály
vhodné na skladovanie energie. Ako možno vidieť na OBR. 1. najväšia
expanzia materiálu sa dosiahne pri prvom teplotnom behu, pričom
opakovaným namáhaním sa materiál rozmerovo stabilizuje. U parafínu S sa
stabilizácia dosiahla už po druhom behu, kdežto pri parafíne FT až po
štvrtom, čo je spôsobené aj intenzitou prechodov medzi tuhou a
kvapalnou fázou, ktorá je výraznejšia u parafínu S.

OBR. 1. Zmena rozmerov zmesi LDPE/ parafín FT (50 / 50 hmot. /
hmot. %) počas termického cyklovania stanovená metódou DMA.
Poďakovanie
Táto práca bola vypracovaná v rámci finačnej podpory projektov
APVV 51-010405 a VEGA 2/6114/26.
Literatúra
1. Zalba B, Marin JM, Cabeza LF, Mehling H, Applied Thermal
Engineering, 23, 251-283 (2003)
2. Zhengguo Zhang, Xiaoming Fang , Energy Conversion and
Management, 47(3), 303-310 (2006)
3. Inaba, H.; Tu, P. Heat Mass Transf 1997, 32, 307-312 (1997)
NEW APPLICATION POSSIBILITIES FOR RECYCLED
POLYOLS
H. BENEŠa, M. DUŠKOVÁa,
M. PEKÁREKa, J. KOVÁŘOVÁa,
J. POSPÍŠILa, K. ZETKOVÁb
aDepartment of Polymer Blends,
Institute of
Macromolecular Chemistry, Academy of Sciences of the Czech Republic,
Heyrovského nám. 2, CZ-162 06 Praha 6, Czech Republic
(benesh(imc.cas.cz)
bSYNPO, a.s., S.K. Neumanna 1316,
CZ-532 07 Pardubice, Czech Republic
Nowadays, the most important method of chemical recycling of
waste
polyurethanes (PUR) is glycolysis. Decomposition of polymer by means of
glycol leads to formation of a product mixture with hydroxyl-end groups
called "recycled polyols". These polyols are suitable for preparation
of new PUR materials, especially rigid PUR foams. Nevertheless, the
rigid PUR foam market is rather limited and thus it is challenging to
find a new application field for these recycled polyols.
We have studied the possibility to use the recycled polyols
from
glycolysis of waste flexible PUR foam for synthesis of PUR coatings.
Coatings were prepared via polyaddition reaction of recycled polyols
with trimer of hexamethylene diisocyanate (t‑HDI) catalyzed by
dibutyltin dilaurate (DBTDL) at room temperature in suitable solvent. A
great advantage of this procedure is usage only recyclate as polyol
component - 100 % replacement of virgin polyol. Two kinds of recycled
polyols (from glycolysis with diethylene glycol- recyclate 1 and
dipropylene glycol- recyclate 2) and acrylic polyol (RCP 18243,
Du-Pont) - for preparation of reference coatings without recyclate -
have been used.
Prepared PUR coatings with recyclates are transparent
slightly
pale-yellow coloured films. Kinetic measurements show sufficient rate
of the polyaddition at room temperature and the reaction is complete
within 24 h. Because of the presence of water in recyclates, the
initial 10‑% molar excess of NCO groups with respect to NCO/ OH
stoichiometry is necessary in order to obtain PUR coating with better
water resistance and higher final hardness. Application time can be
adjusted by adding suitable amount of DBTDL. Appropriate application
time around 2 h is achieved with addition of 200 ppm DBTDL. Content of
solvent in the reaction system is driven by environmental issue leading
to the reduction of VOCs from the coatings (according to Solvents
Emission Directive 1999/13/EC). From this point of view, an effort is
to prepare the reactive PUR coatings with low content of solvents (i.e.
"high solids" coatings with content of solvent lower than 40 wt.%). The
usage of "slow" solvents (with high boiling point - above 150°C) is
preferred due to their slower evaporation rate avoiding formation of
opalescent film due to the phase separation or reaction with air
humidity. 2-Heptanone or bis(2-methoxyethyl)ether seem to be suitable
solvents.
Obtained PUR coatings with recyclates are thermally stable up
to
200°C. Because of the presence of long aliphatic chain and flexible
ether bonds, the PUR coatings with recyclates are very flexible and
have superior mechanical properties, like bending, impact resistance,
and cupping. Drying times of all PUR coatings are at the normal level,
as well as hardness. Adhesion to various substrates was tested; good
adhesion is to glass, steel and PUR top coat substrates, bad adhesion
is to aluminium and zinc substrates (adhesion is improved by oxidizing
of zinc substrate). The adhesion becomes rapidly worse after the
exposure the coatings to water (also in the case of reference coating
without recyclate).
Because of the presence of the aromatic part in the recyclates
(MDI
from original flexible PUR foam), which has a strong tendency to
yellowing presumably by photooxidation, the test of photodegradation
(QUV accelerated ageing in weatherometer) of the PUR coatings with
recyclate components was also performed. First results show, that after
500 h exposure no significant changes in IR spectra, indicating an
early stage of photodegradation process, were observed.
Acknowledgement. Financial support by grant
SL-7-26-05
from the Ministry of Environment of the Czech Republic is gratefully
appreciated.
Rh COMPLEXES ANCHORED ON INORGANIC AND
POLYMERIC
SUPPORTS: HETEROGENEOUS CATALYSTS FOR POLYMERIZATION OF PHENYLACETYLENE
AND ITS RING-SUBSTITUTED DERIVATIVES INTO HIGH-PURITY POLYMERS
SVOBODA J., SEDLÁČEK J., RÉDROVÁ D., DVOŘÁKOVÁ G., VOHLÍDAL J.
Department of Physical and Macromolecular Chemistry,
Laboratory
of Specialty Polymers, Faculty of Science, Charles University, Albertov
2030, CZ-128 40, Prague 2, Czech Republic
(js(vivien.natur.cuni.cz)
Introduction
Rh(I) complexes are for two decades known as efficient
catalysts for
homogeneous insertion polymerization of substituted acetylenes into
stereoregular polyacetylenes, i.e. into the specialty polymers with
potential application in electronics and optoelectronics. If
polyacetylenes are prepared with homogeneous catalysts the catalyst
residues buried in the polymers deteriorate functional properties of
polymers prepared since they act as dopants or as quenchers of excited
states or traps for charge carriers. Catalyst residues are mostly
removed from polymers by repeated precipitation or by adsorption that
are time consuming and limited efficiency processes. Therefore, a
development of heterogeneous polymerization catalysts is highly
desirable, since they can be easy separated from the reaction mixture,
which opens a way to high-purity polymers. In this contribution a
survey is given on our recent results in the field of heterogeneous
catalysts for acetylenes polymerization prepared by anchoring Rh(I)
complexes on mesoporous inorganic and polymeric supports.
Results and Discussion
All siliceous molecular sieves MCM-41, MCM-48 and SBA-15
porous
polybenzimidazole (PBI) (Hoechst-Celanese Corporation), and
(aminomethyl)polystyrene-co-divinylbenzene (AMPS-DVB) (Aldrich) were
used as supports for anchoring homogeneously active Rh(I) complexes.
[Rh(COD)OCH3]2 and
[Rh(cyclodiene)acac]
(cyclodiene = cycloocta-1,5-diene (COD) or norbornadiene (NBD), acac =
acetylacetonato) were anchored on molecular sieves via ligand exchange
reaction with sieves OH surface groups under elimination of CH3OH
and acetylacetone, respectively. For anchoring [Rh(cyclodiene)Cl]2
complexes molecular sieves have been modified by NH2(CH2)3Si(OCH3)3
(APTMS) or PPh2(CH2)2Si(OC2H5)3
(DPPETES) linkers to introduce ligands that coordinate well to Rh
species. Sieves modification is accompanied by the liberation of
corresponding alcohohol into the liquid phase. On the contrary,
[Rh(cyclodiene)Cl]2 complexes were easy anchored
on
non-modified PBI and AMPS-DVB. ESCA analysis of the heterogeneous
catalysts has shown that chlorine atoms remain bound to the anchored Rh
species. This indicates that, during anchoring, dinuclear Rh species
are transformed into the mononuclear ones which are then bound onto the
support via the nitrogen groups. Attempts to anchor [Rh(COD)OCH3]2
and [Rh(cyclodiene)acac] complexes on PBI and AMPS-DVB were
unsuccessful.
Scheme 1 Polymerization
of phenylacetylene and its ring-substituted derivatives with Rh
heterogeneous catalysts.
In all cases, the anchoring was accomplished by the direct
adsorption of the complex from CH2Cl2
or THF solution, the course of anchoring was monitored by UV/vis
spectroscopy. In addition, the content of Rh in the resulting catalysts
was determined by ICP-MS. Heterogeneous catalysts with 0.3 - 3 wt.%
Rh-loading were prepared by this approach.
The polymerizations of substituted acetylenes [phenylacetylene
(PhA), its 4-pentyl-, 2-F-, 4-F- and 4-(Me3Si-CºC-C6H4CH=N-)
derivatives] (Scheme 1) were performed in batch reactor using SEC for
monitoring the course of reaction and ICP-MS for monotirong catalyst
leaching. In addition, standard filtrate tests were performed in the
course of polymerization that confirmed that reaction proceeds in the
system with the heterogeneous catalyst only and not in withdrawn
filtrate.
Rh(COD) complexes anchored on PBI, non-modified molecular
sieves,
and molecular sieves modified with APTMS linker exhibit good activity
in polymerization of PhA and its dereivatives (Tab. 1). Polyacetylenes
prepared are of the same high cis-transoid configuration as those
resulting with corresponding homogeneous catalysts. Catalysts give easy
separable high-purity neat polymers containing from 0.00002 to 0.0001
wt.% of Rh, which represents 0.002 - 0.015 % of the catalyst used.
Reference polymers prepared by homogeneous processes contain from 0.01
- 0.1 wt.% of Rh (1 - 15 % of the catalyst used). This means that the
transformation of homogeneous processes to heterogeneous ones brings
about an approx. thousand-fold reduction in neat polymer contamination
with Rh residues.
Table 1 - Yield (Y) and weight-average
molecular weight (Mw) of poly(PhA) achieved with
catalysts based on [Rh(COD)Cl]2. [PhA]0
= 0.6 M, [Rh] = 1.5 mM; THF, room temperature, 7 h.
|
Support
|
Linker
|
Y(%)
|
10-3Mw
|
|
MCM-41, MCM-48, SBA-15
|
APTMS
|
43 - 52
|
110-190
|
|
PBI
|
-
|
50a)
|
140a)
|
|
MCM-41
|
DPPETES
|
15
|
5,6
|
|
AMPS-DVB
|
-
|
5
|
56
|
|
b)
|
b)
|
65b)
|
88b)
|
a) [PhA]0 = 1.2 M
b) homogeneously catalysed with
[Rh(COD)Cl]2
Rh(COD) complex anchored on molecular sieves modified with
DPPETES
is less effective polymerization catalyst, most probably owing to too
high concentration of polymerization disturbing -PPh2
groups
in the vicinity of the active sites. With Rh(COD) anchored on AMPS-DVB
the polymerization in very rapid in the initial stage, however, polymer
formed is most probably insufficiently rapidly released from the
catalyst pores and reaction is slowed down and subsequently stopped.
The above-described successful results obtained with anchored
Rh(COD) complexes have not been achieved with Rh(NBD) complexes, which
provide considerably lower yields of polyacetylenes. This can be
explained by the fact that Rh(NBD) complexes are known to induce
polymerization with restricted extent of kinetic-chain transfer (even
of living or living like type). Therefore, it seems to be that the
formed polymer chains remain bound to active polymerization centers,
and thus they cannot be released from the catalyst pores into the
surrounding solution and most probably partly block the active sites of
polymerization.
Acknowledgement:
Financial support form the Grant Agency of the Czech Republic
(project No. 203/05/2194) is gratefully acknowledged. Co-author Jan Svoboda is indebted to the
Grant Agency of the Czech Republic for the fellowship (project No.
203/03/H140).
Preparation and application of dinuclear
carboxylate bridged rhodium diene complexes
J. ZEDNÍK, J. SVOBODA, J. SEDLÁČEK, J. VOHLÍDAL and D. RÉDROVÁ
Department of Physical and Macromolecular Chemistry,
Laboratory of
Specialty Polymers, Charles University, Albertov 2030, CZ-128 40,
Prague 2, Czech Republic; E-mail: jzednik(centrum.cz
At the present time rhodium complexes attract great interest
in
many research groups. Since they can catalyze many different chemical
reactions such as hydroformylation, hydrosilylation, hydrogenation and
polymerization of dienes, acetylenes and some other monomers. Rhodium
based catalysts are highly tolerable to reaction conditions, solvents
(alcohol, water, ionic liquids, etc.) and functional groups of
reactants. Further advantage is the possibility to anchor rhodium
complexes on various types of heterogeneous support such as porous
polybenzimidazole beads, polystyrene matrices and mesoporous molecular
sieves to form hybrid catalysts that could be easily separated from the
reaction media.

Fig. 1. Rhodium complexes prepared
In this contribution we refer to catalytic applicability of
diene
carboxylate bridged Rh complexes, which were synthesized in our group.
Carboxylate complexes (I-VIII) are highly active in polymerization of
phenylacetylene if tetrahydrofuran (THF) is used as solvent (Tab. 1).
In opposite all complexes exhibit only oligomerization and
cyclotrimerization activity when non-polar solvents such as
dichloromethane (DCM) are used.
Tab. 1. Comparison of activity of different
complexes in
polymerization of phenylacetylne in tetrahydrofurane (THF) and
dichloromethane (DCM), initial monomer concentration, [M]0
= 0.6 mol.l-1, initial monomer to Rh mole ratio,
[M]0/[Rh] = 200, reaction time = 420 min, room
temperature, YO -
yield of oligomers, YP -
yield of polymer, Mn and Mw
is the number-average and weight average molecular weight of polymer
relative to polystyrene standards.
|
THF
|
DCM
|
|
complex
|
YO, %
|
YP, %
|
10-3.Mn
|
Mw/Mn
|
YO, %
|
YP, %
|
|
I
|
7
|
16
|
28
|
2,4
|
14
|
0
|
|
II
|
14
|
15
|
7
|
2,1
|
47
|
0
|
|
III
|
16
|
21
|
20
|
2,6
|
24
|
0
|
|
IV
|
22
|
20
|
19
|
3,2
|
22
|
0
|
|
V
|
21
|
29
|
11
|
2,4
|
26
|
0
|
|
VI
|
23
|
23
|
14
|
2,2
|
22
|
0
|
|
VII
|
25
|
25
|
27
|
3,0
|
19
|
0
|
|
VIII
|
21
|
23
|
12
|
2,3
|
27
|
0
|
Complex (I) was used also for atom transfer ATRP
polymerization of
styrene a and methyl metacrylate. In toluene in presence of Bu2NH
(monomer/Rh mole ratio 800) complex exhibit first order kinetic for
both monomers (polystyrene a and methyl metacrylate). Addition of Bu2NH
in relation to Rh increased the polymer yield, decreased the polymer
molecular weight and significantly increased initiator efficiency in
all polymerization systems tested.
Tab. 2. Results of ATRP
polymerization styrene (sty) and methyl metacrylate (MMA) using complex
(I) as a catalyst.
|
Monomer
|
[M]/[Rh]
|
Yp, %
|
Mna
x 10-3
|
Mw/Mna
|
fb
|
|
MMA
|
100
|
39
|
26
|
1.50
|
0.08
|
|
MMA
|
800
|
38
|
397
|
1.45
|
0.04
|
|
MMA + Bu2NHc
|
800
|
95
|
116
|
1.46
|
0.33
|
|
Sty
|
100
|
56
|
41
|
1.67
|
0.07
|
|
Sty
|
800
|
23
|
126
|
1.55
|
0.08
|
|
Sty + Bu2NHc
|
800
|
80
|
39
|
1.27
|
0.85
|
a) Mn determined
by SEC using poly(methyl metacrylate) calibration for PMMA and
polystyrene calibration for PS. b) Initiator
efficiency: f = Mn, theor/Mn,
exp where Mn, theor = ([M]0/[I]0)Mmon
Y([M]0) and [I]0 is
initial concentration of monomer and initiator, respectively, Mmon
is molecular weight of monomer), c) Four
equivalents of Bu2NH with respect to Rh
Complexes (V) and (VI) were tested for hydroformylation of
1-octene
and styrene. Complexes exhibit activity with good regioselectivity, but
reaction rates are low, probably due to steric effects exerted by their
bulky carboxylate ligands.
Fig. 2. Hydroformylation of 1-octene using
carboxylate rhodium catalysts
Tab. 3. Hydroformylation of styrene and
1-octene using
rhodium carboxylate complexes V and VI. Reaction conditions: Catalysis
was performed by a 0.1 mM solution of corresponding rhodium complex in
CH2Cl2;
substrate:catalyst = 1000:1; initial total pressure of CO/H2
(1:1) = 1000 psi.
|
substrate
|
Complex
|
T (°C)
|
Time (h)
|
Conversion (%)
|
RBr (%)
|
Selectivity
2:3:4:5
|
TON
|
|
Styrene
|
V
|
25
|
48
|
17
|
97
|
-
|
170
|
|
Styrene
|
V
|
60
|
16
|
40
|
97
|
-
|
400
|
|
Styrene
|
VI
|
25
|
48
|
12
|
97
|
-
|
120
|
|
Styrene
|
VI
|
60
|
16
|
38
|
93
|
-
|
380
|
|
1-octene
|
V
|
25
|
48
|
76
|
-
|
54:46:-:-
|
760
|
|
1-octene
|
V
|
60
|
16
|
100
|
-
|
52:37:8:3
|
1000
|
|
1-octene
|
VI
|
25
|
48
|
63
|
-
|
55:45:-:-
|
630
|
|
1-octene
|
VI
|
60
|
16
|
100
|
-
|
52:41:6:1
|
1000
|
(Turnover no. - TON ) = aldehydes fraction x
substrate:catalyst ratio, Rbr = regioselectivity
towards branched aldehydes.
References:
- Sedláček J., Vohlídal J.: Collect. Czech. Chem.
Commun. 2003, 68,
1745.
- Opstal T., Zedník J., Sedláček J., Svoboda J., Vohlídal J.,
Verpoort F.: Collect. Czech. Chem. Commun. 2002,
67, 1858.
- Kostas I. D., Vallianatou K. A., Kyritsis P., Zedník J.,
Vohlídal J.: Inorg. Chim. Acta 2004,
357, 3084.
Mechanistic investigation of non-MAO
activated nickel
diimine catalyzed olefin polymerization by means of UV-visible
spectroscopy
J. Peleškaa, S. Hermanováa,
J. MERNAb*
aInstitute of Material Science, Brno
University of Technology, Purkyňova 118, CZ-612 00 Brno
bDepartment of Polymers, Institute of
Chemical Technology Prague, Technická 5, CZ-166 28 Praha 6,
jan.merna(vscht.cz
UV-VIS spectroscopy has been shown to be effecient metod for
evaluation of basic mechanistic steps in both early and late metal
complexes catalyzed olefin poylmerization1-4.
UV-visible spectra of
(N,N'-bis(2-t-butylphenyl)-1,8-naftalendiyl-1,4-diazabuta-1,3-diene)nickeldibromide
(Ni1, Fig. 1) activated by methylalumoxane (MAO)
show always two intensive and broad absorption bands3,5.
These were tentatively attributed to active (530 nm) and inactive (700
nm) species.In this contribution we focus on UV-visible spectra of Ni1
activated by simple organoaluminium compounds.
Our recent investigations have shown that trialkylaluminium,
dialkylmagnesium, alkyllithium and dialkylzinc compounds are
inefficient cocatalyst for complex Ni1 in propylene
and hex-1-ene polymerization. On the other hand very active catalytic
system is obtained upon activation of Ni1 by
diethylaluminiumchloride (DEAC) and ethylaluminiumdichloride (EADC)6.
In contrast with Ni1/MAO system, both Ni1/DEAC
and Ni1/EADC
absorption spectra show only one strong peak centered at 530 nm in the
presence of hex-1-ene. This suggests that in the beginning of reaction
there is only one type of active centers present in the system. As the
system is active in polymerization we believe the peak belongs to
alkyl-olefin complex recognized by Brookhart as a resting state of
catalyst (Fig.1B)7. Further, changes of spectra
in course of
the polymerization at different reaction conditions will be given. The
importance of order of components addition will be discussed as well.

Fig. 1 Structure of nickel diimine catalyst Ni1
(A) and the structure of proposed catalytically
active specie upon activation with DEAC (B)
1. Pedeutour, J.-N.; Coevoet, D.; Cramail, H.; Deffieux, A. Macromol.
Chem. Phys. 1999, 200,
1215.
2. Luo, H.-K.; Yang, Z.-H.; Mao, B.-Q.; Yu, D.-S.; Tang, R.-G.
J. Mol. Cat. A: Chemical 2002, 177,
195.
3. Puruch, F.; Cramail, H.; Defieux, A. Macromolecules
1999, 32, 7977
4. Simon, L. C.; Williams, C. P.; Soares, J. B. P.; Souza, R.
F. J. Mol. Cat. A: Chemical 2001,
165, 55.
5. Merna, J., PhD Thesis, Brno University of Technology, 2005
6. Peleska, J., Thesis, Brno University of Technology, 2006
7. Johnson, L. K.; Killian, C. M.; Brookhart, M. J.
Am. Chem. Soc. 1995, 117,
6414.
ASSOCIATION BEHAVIOR OF HETEROARM STAR
COPOLYMER
A MONTE CARLO STUDY
J. HAVRÁNKOVÁ, Z. LIMPOUCHOVÁ, K. PROCHÁZKA
Department of Physical and Macromolecular Chemistry, Faculty
of
Science, Charles University, Albertov 6, 128 43 Prague 2, Czech Republic
Association behavior of heteroarm star copolymers with equal
number
of different arms was studied by dynamic lattice Monte Carlo
simulations. We have investigated the influence of star architecture
(number and length of arms) and quality of selective solvent on
formation and structure of associates. Simulation yield very detailed
information on the system at molecular level and showed differences in
core-shell structure of associates creating by linear diblocks and by
star copolymer.
CONFORMATIONAL BEHAVIOUR OF COMB-LIKE
POLYELECTROLYTES IN SELECTIVE SOLVENT; COMPUTER SIMULATION STUDY.
P. KOŠOVANa, Z. LIMPOUCHOVÁa,
K. PROCHÁZKAa
aDepartment of Physical and
Macromolecular Chemistry,
Faculty of Science, Charles University in Prague, Albertov 6, 128 43
Praha 2, Czech Republic (kosovan(vivien.natur.cuni.cz)
Introduction
Conformational behaviour of
polyelectrolytes is a result of interplay of electrostatic repulsion
between charged monomer units on one hand and hydrophobic attraction on
the other hand. This results in a variety of conformational structures.
In linear polyelectrolytes in poor solvent, so-called pearl-necklace
structures are formed [1] [2].
The behaviour of comb-like
amphiphilic
graft copolymers has been studied recently by Borisov and Zhulina [3].
They have shown that they exhibit a series of conformational
transitions in a solvent, which is poor for the backbone and good for
the grafts.
Behaviour similar to that
reported by
Borisov and Zhulina might be expected for a comb-like copolymer with a
hydrophobic backbone, grafted with polyelectrolyte side chains for
which the solvent is only moderately poor. Then the actual
conformational behaviour will depend on the interplay between the
degree of charging of the side chains and their hydrophobicity. Our
current study explores how the degree of charging of the side chains
and topology of the comb polyelectrolyte affect the conformation
Polymer model
We use molecular dynamics
simulations with Langevin thermostat, implicit solvent and explicit
electrostatic interactions.
The model polymer has strongly
hydrophobic
neutral backbone, grafted with moderately hydrophobic polyelectrolyte
side chains. The interaction parameters of the model roughly correspond
to PS-graft-PMA. The length of the backbone is 60, 120 and 240 monomer
units and the total number of monomer units of the grafts is 600. The
number and the length of the grafts is varied from 10 grafts of 60
monomer units each (10x60) through 30x20 up to 60x10.
Results
We have studied the effect of
grafting and
degree of dissociation on the conformational behaviour Figure 1 shows
simulation snapshots of polymers of different molecular architectures
and various degree of dissociation of the grafts. The leftmost column
shows polymers with 10 grafts, each 60 monomer units long (10x60).
Architectures of 30x20 and 60x10 follow in the next two columns.
Vertically three different degrees of dissociation (0.125, 0.25 and
0.50 respectively) are shown. Below the snapshots we plot the average
values of bond angle cosines within the backbone (denoted by 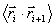 ,
where
,
where 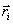 is the bond vector from monomer i
to monomer i+1)
as functions of monomer position in the backbone. The bond angle
cosines average out to 0.0 for a random coil and approach the value of
1.0 for a string. For intermediate degrees of dissociation alternating
strings and pearls can be observed along the backbone (indicated by one
or more "bumps" in the corresponding plot of bond angle cosines).
is the bond vector from monomer i
to monomer i+1)
as functions of monomer position in the backbone. The bond angle
cosines average out to 0.0 for a random coil and approach the value of
1.0 for a string. For intermediate degrees of dissociation alternating
strings and pearls can be observed along the backbone (indicated by one
or more "bumps" in the corresponding plot of bond angle cosines).
With all three type of polymer
architecture
the conformation expands with increasing dissociation of the grafts. In
a polymer with few long grafts, the increase in electrostatic repulsion
is compensated for by extending the long side chains. In the other two
cases the grafts are too short and the expansion proceeds through
formation of pearl-necklace structures, similar to those in linear
polyelectrolytes. Unlike in the case of linear polyelectrolytes they
are stable, what can be seen from the plots of bond angle cosines.
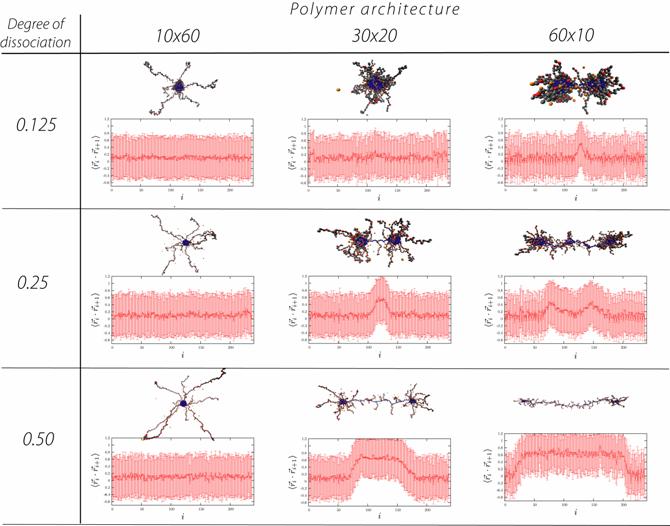
Fig. 1 Simulation snapshots and plots for
various polymer
architectures (horizontal) and degrees of dissociation (vertical). The
plots of bond angle cosines below the corresponding snapshots confirm
the presence of pearl-necklace structures.
Conclusions
Our results show that the
comb-like
polyelectrolytes form self-organized pearl-necklace structures under
suitable conditions. Unlike in linear polyelectrolytes the
pearl-necklace structures are stable with respect to fluctuations and
can be visualised by plotting the bond-angle cosines.
The behaviour can be
controlled by the
degree of dissociation of the grafts and ionic strength of the
solution, while it is independent of polymer concentration as long as
the solution is dilute. If the grafts comprise a weak polyelectrolyte
(e.g. PMA) the conformational behaviour can be easily controlled by
solution pH.
This behaviour is
qualitatively similar to
what was predicted theoretically by Borisov and Zhulina for neutral
amphiphilic graft copolymers [1].
References
1 A.V. Dobrynin, M. Rubinstein, S.P.
Obukhov; Macromolecules 1996, 29,
2974
2 H.J. Limbach, C. Holm; J.
Chem. Phys. 2001, 114, 9674
3 O.V. Borisov; E.B. Zhulina; Macromolecules
2005, 38, 2506.
Kvapalinová chromatografia polymérov pri limitných podmienkach entalpických interakcií.
D. Berek
Ústav polymérov SAV, 84236 Bratislava, SK, E-mail dusan.berek(savba.sk
V príspevku opíšeme nový prístup k separácii polymérov, založený na
rozdielnej dynamike transportu malých a veľkých molekúl v
chromatografickej kolóne. Nazývame ho kvapalinovou chromatografiu pri
limitných podmienkach entalpických interakcií (liquid chromatography
under limiting conditions - LC LC). Malé molekuly eluenta alebo vhodnej
pomocnej látky vnikajú do všetkých pórov náplne kolóny a v dôsledku
toho je ich transport spomaľovaný. Naopak, makromolekuly, ktoré sú
čiastočne alebo úplne vylúčené z pórov náplne, majú tendenciu pohybovať
sa kolónou rýchlo. Je možné nájsť také experimentálne podmienky, pri
ktorých malé molekuly vytvoria „bariéru“, zamedzujúcu rýchly pohyb
makromolekúl. Využiť možno akúkoľvek kontrolovanú entalpickú
interakciu; dosiaľ sme otestovali adsorpciu, entalpickú particionáciu i
fázovú separáciu, makromolekúl. Podľa potreby teda bariéra malých
molekúl podporuje adsorpciu alebo entalpickú particionáciu makromolekúl
na vhodnej náplni kolóny, prípadne bariérou je zrážadlo pre polymér.
Bariéra môže pracovať selektívne, t.j. blokovať rýchly transport len
jedného typu makromolekúl, zatiaľ čo iný typ makromolekúl sa bude
vymývať bez prekážky. Tak možno veľmi rýchlo a bezpečne oddeliť
polyméry s rôznym chemickým zložením alebo architektúrou. Bariérou môže
byť samotná mobilná fáza – vtedy sa vzorka rozpustí a dávkuje do kolóny
v rozpúšťadle, ktoré podporuje jej voľné vymývanie. Makromolekuly so
sklonom k entalpickým interakciám s náplňou kolóny alebo také, pre
ktoré je eluent zrážadlom, nemôžu opustiť zónu svojho pôvodného
rozpúšťadla. Výhodnejší sa javí prístup, pri ktorom sa úzka zóna
kvapaliny brániaca rýchlemu transportu makromolekúl, teda bariéra,
dávkuje bezprostredne pred vzorkou alebo spolu s ňou. V oboch prípadoch
sa „zabrzdené“ makromolekuly akumulujú na hrane bariéry a vymyjú sa z
kolóny v podobe úzkeho, fokusovaného píku - a to bez ohľadu na ich
mólovú hmotnosť. Fokusácia chromatografickej zóny vzorky patrí medzi
významné prednosti LC LC. Umožňuje nadávkovanie veľkého objemu vzorky
do kolóny. Ukázalo sa tiež, že LC LC systémy sú zväčša veľmi robustné,
pracujú v širokom rozmedzí zložení mobilných fáz a teplôt. Ďalšími
prednosťami LC LC metód je ich aplikovateľnosť na vzorky s veľmi
vysokou mólovou hmotnosťou a veľká zaťažiteľnosť kolón. To umožňuje
separáciu a nasledujúcu on-line GPC molekulovú charakterizáciu
minoritných (< 1%) zložiek zmesí polymérov, ktorá je pomocou
konvenčných metód prakticky neuskutočniteľná. Separácia makromolekúl
rôzneho zloženia alebo architektúry sa dá vykonať veľmi rýchlo, v
priebehu dvoch – troch minút. Zásadná prednosť LC LC s úzkou zónou
rozpúšťadla podporujúceho adsorpciu polyméru je prakticky úplná
výťažnosť vzoriek vo všetkých dosiaľ testovaných systémoch. Ukazuje sa,
že LC LC bude možné využiť i na separáciu štatistických kopolymérov a
stereoregulárnych polymérov.